The Digital Health Revolution is happening all around us and it is both exciting and terrifying. As a member of the Medical Device community, we have a responsibility to our patients, our families, and our shareholders to follow the current best practices. To date, the standards and regulations have not been able to keep pace with the needs of the patients and the industry that satisfies these patient and user needs.
Topics in this newsletter:
Artificial Intelligence regulation
Many regulatory agencies including the FDA are talking about forming committees that will codify the “current best practice”, often referred to as GxP or “Good [insert name] Practice”. In January 2020, the FDA released a discussion paper titled US FDA Artificial Intelligence and Machine Learning Discussion Paper, in this paper, the agency proposes a framework for Good Machine Learning Practices, and below is a visual representation of the GMLP workflow.
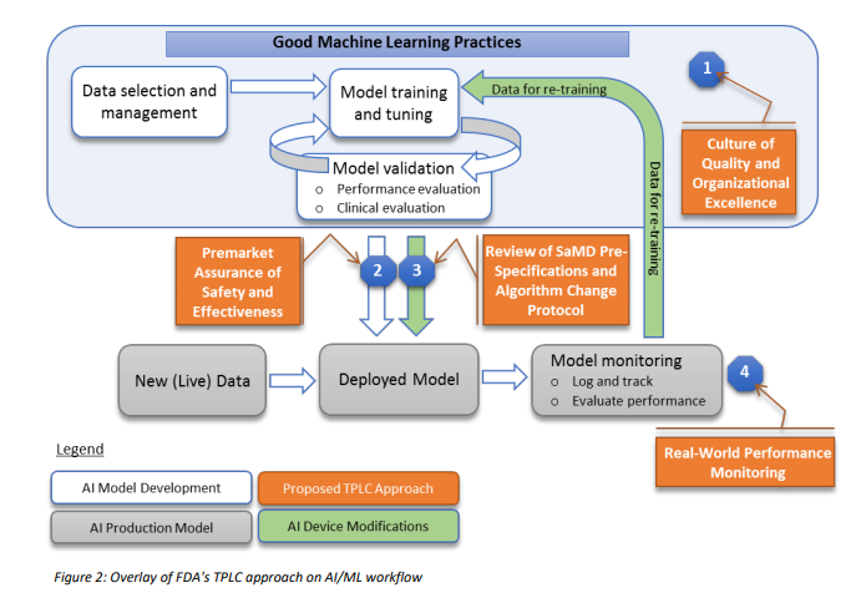
In addition, to address the critical question of when a continuously learning AI/ML SaMD may require a premarket submission for an algorithm change, this discussion paper proposes a framework for modifications to AI/ML-based SaMD.
To date, the FDA has cleared or approved several AI/ML-based SaMD however these have only included algorithms that are “locked”. The power of many of these AI/ML-based SaMD lies within the ability to continuously learn, where the adaptation or change to the algorithm is realized after the SaMD is distributed for use and has “learned” from real-world experience. Following the distribution, these types of continuously learning and adaptive AI/ML algorithms may provide a different output in comparison to the output initially cleared for a given set of inputs.
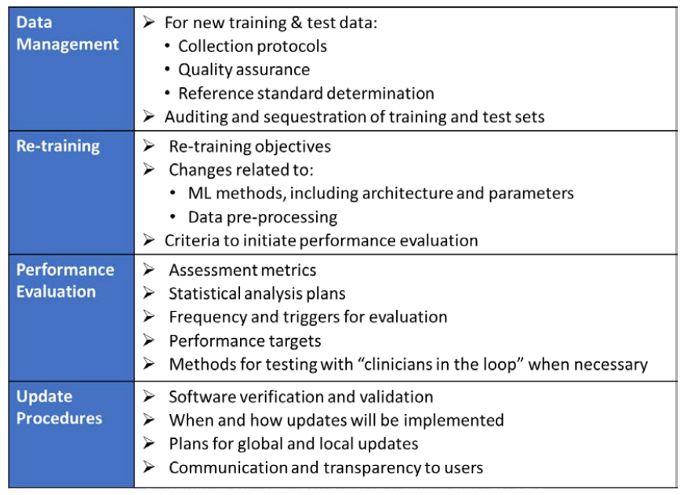
To adapt to this, the FDA is proposing a principle of a “predetermined change control plan.” The predetermined change control plan would include the types of anticipated modifications based on the retraining and model updating strategy, and the associated methodology – referred to as the Algorithm Change Protocol (ACP) – to be used to implement those changes in a controlled manner that manages risks to patients, see below for an outline of the main components of an ACP:
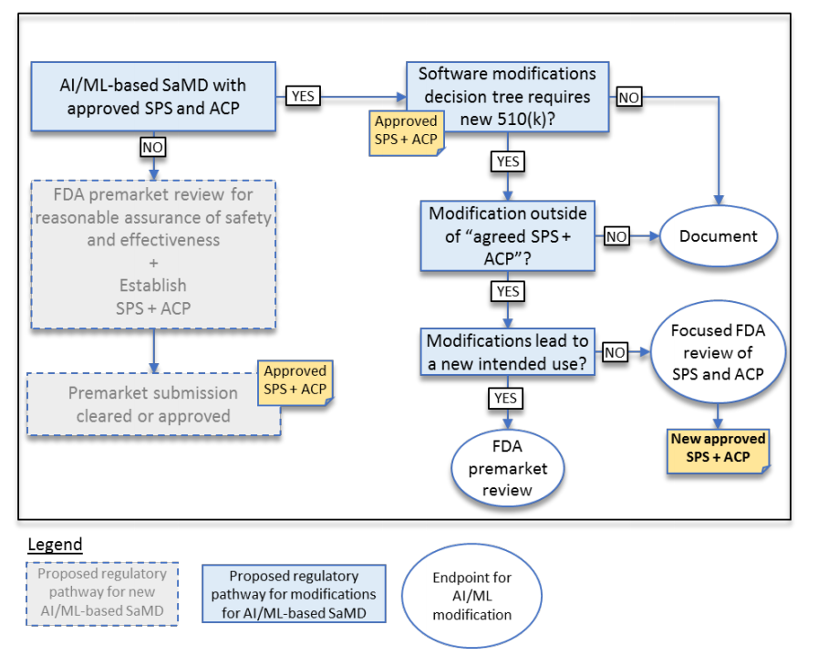
The result is a modification to the current guidance for software Deciding When to Submit a 510(k) for a Software Change to an Existing Device | FDA. See below for a proposal to the current guidance
Multiple functions – one device
Back in July 2020, the FDA released a Final Guidance Document looking at what happens when the same medical product has both functions covered by the FDA and functions not covered by the FDA. The uniquely unclear guidance titled Multiple Function Device Products: Policy and Considerations look at what happens when a single product with multiple functions have some that are “regulated” and require FDA review, clearance, or approval other functions that do not require FDA involvement (other regulations like FCC or CE standards for electronics, RoHS, WEEE, UL and other TLA’s may be required).
A “function” is a distinct purpose of the product, which could be the intended use or a subset of the intended use of the product. A product with an intended use is to store, transfer, and analyze data has three functions: (1) storage, (2) transfer, and (3) analysis.
While storage & transfer may not be considered requiring regulatory oversight, the addition of analysis and the type of analysis may require FDA involvement. To make things even more complicated, the FDA has issued guidance that indicates while not “fully ok” the FDA does not intend to focus its regulatory oversight on some devices that pose a low risk to patients for more on this ever-growing category see the FDA’s guidance “Policy for Device Software Functions and Mobile Medical Applications” and “General Wellness: Policy for Low-Risk Devices.”
Many CEO’s will attempt to push themselves into this “low-risk category”, as with most things, the agency has discretion, but with software being the leading cause of recalls in the US, the FDA will be waiting to investigate any complaints and will be looking to punish those that have not followed the guidelines to the agencies liking.
So how do we determine if the “non-FDA-regulated” or as the FDA likes to call it the “other function” impacts on the “regulated” feature?
Start with 2 questions, and answer them as if you were working for the FDA:
1) Is there an impact on the safety or effectiveness of the “regulated” feature as a result of the “other function?”
if yes,
2) Could the impact result in increased risk or have an adverse effect on the performance of the device function-under-review.
I would very much like to say that from the FDA's perspective the following is true:
- if the “other function” shares code then the answer to both is yes.
- if the “other function” shares the same output screen or graphical user interface, the answer to both is yes.
However, there are always exceptions and those edge cases are why we sometimes need to speak with a member of the Gsap digital health team Regulatory Review Team to confirm.
Below are a number of relevant examples from the guidance for your consideration, while some may enlighten, others may confuse, but that is the art of regulatory science.
This Newsletter Prepared by:

Yaron Eshel, Q&R project manager
Medical device, Digital Health Discussion Team
For more information about our Digital Health services visit: