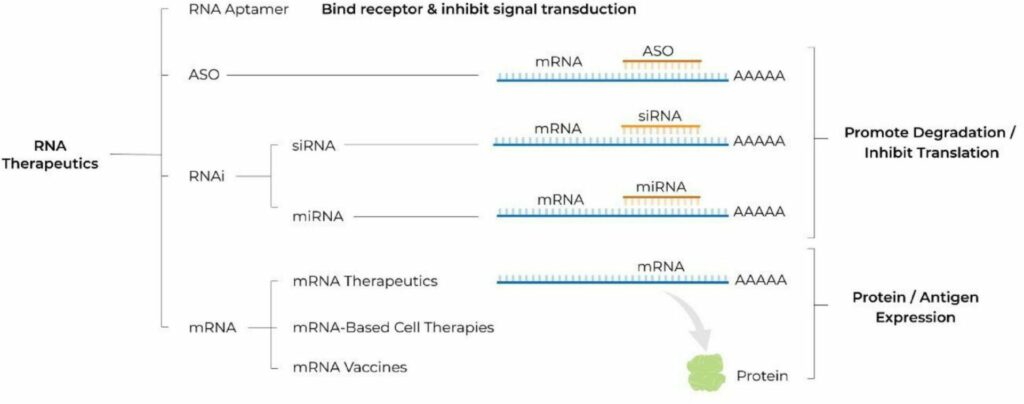
The recent acquisition of V-Wave by Johnson & Johnson marks a significant milestone in the medical device industry. At the core of this acquisition is V-Wave’s innovative interatrial shunt, a device designed to address the needs of heart failure patients. However, what makes this story truly remarkable is V-Wave’s successful navigation of the FDA’s Breakthrough Devices Program. This regulatory pathway played a crucial role in accelerating the device’s journey to market.
The FDA Breakthrough Devices Program: An Overview
Origins and Purpose
The FDA’s Breakthrough Devices Program was established under the 21st Century Cures Act, signed into law in December 2016. The program’s primary objective is to expedite the development and review of medical devices that offer more effective treatment or diagnosis for life-threatening or irreversibly debilitating conditions. By fast-tracking the approval process, the program aims to provide patients and healthcare providers with timely access to innovative medical technologies while upholding the FDA’s rigorous standards for safety and efficacy.
Eligibility Criteria for the Breakthrough Devices Program
To be eligible for the Breakthrough Devices Program, a device must meet the following criteria as outlined by the FDA:
- It provides for more effective treatment or diagnosis of life-threatening or irreversibly debilitating human diseases or conditions; and
- It meets at least one of the following criteria:
- Represents Breakthrough Technology;
- No approved or cleared alternatives exist;
- Offers significant advantages over existing approved or cleared alternatives, including the potential, when clinically compared to existing approved or cleared alternatives, to reduce or eliminate the need for hospitalization, improve patient quality of life, facilitate patients’ ability to manage their own care (e.g., through self-directed personal assistance), or establish long-term clinical efficiencies;
- Device availability is in the best interest of patients.
These criteria are stringent and require substantial evidence. Companies must demonstrate that their device addresses a significant unmet medical need and has the potential to make a meaningful impact on patient outcomes. The rigorous nature of these criteria is reflected in the selectivity of the program, as only devices that show promise in significantly advancing patient care are granted designation.
Key Statistics and Program Impact
Since its inception, the Breakthrough Devices Program has granted designation to over 700 devices. However, it’s important to note that not all devices receiving this designation ultimately gain FDA approval. The program is highly selective, and only those devices demonstrating significant clinical benefits are likely to succeed.
The growth in the number of granted designations over the years is indicative of the increasing recognition and utilization of the program by the medical device industry. This growth is illustrated in the following graph, which shows the number of Breakthrough Device designations granted by fiscal year:
Figure 1: Number of Granted Breakthrough Device Designations by Year
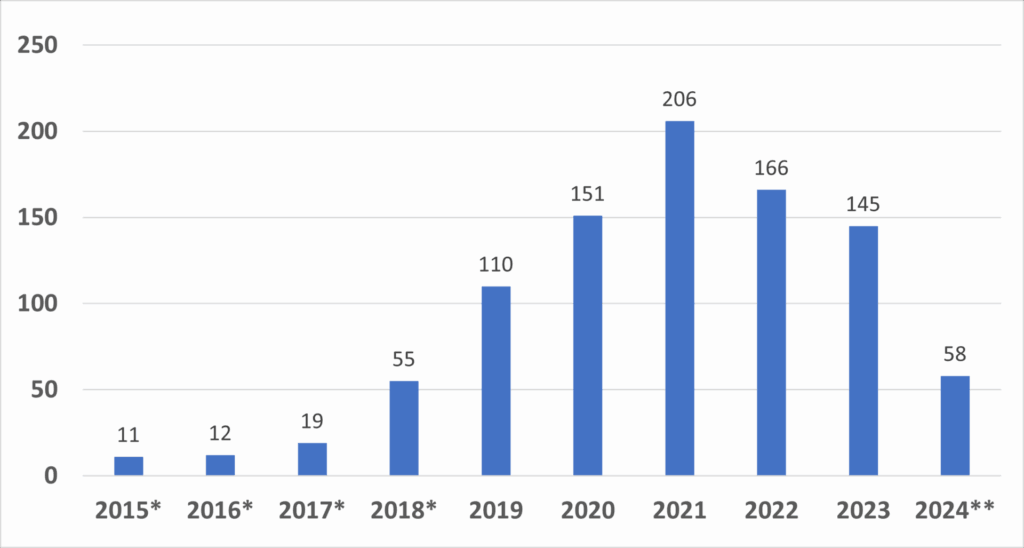
Data includes devices designated under the precursor Expedited Access Pathway (EAP). Since the vision and designation criteria between the precursor EAP Program and the Breakthrough Devices Program are consistent, the FDA considers devices granted designation under the EAP to be a part of the Breakthrough Devices Program.
*Indicates the 2024 data are from October 1, 2023, through December 31, 2023.
Source: FDA
This graph demonstrates the program’s growing impact, with a noticeable increase in designations from 2018 onward. This surge is a testament to the program’s importance in accelerating the development of innovative medical technologies, particularly those targeting unmet medical needs.
Furthermore, the diversity of clinical panels benefiting from the program underscores its broad applicability across various medical fields. The following graph details the number of Breakthrough Device designations by clinical panel:
Graph 2: Number of Granted Breakthrough Device Designations by Clinical Panel
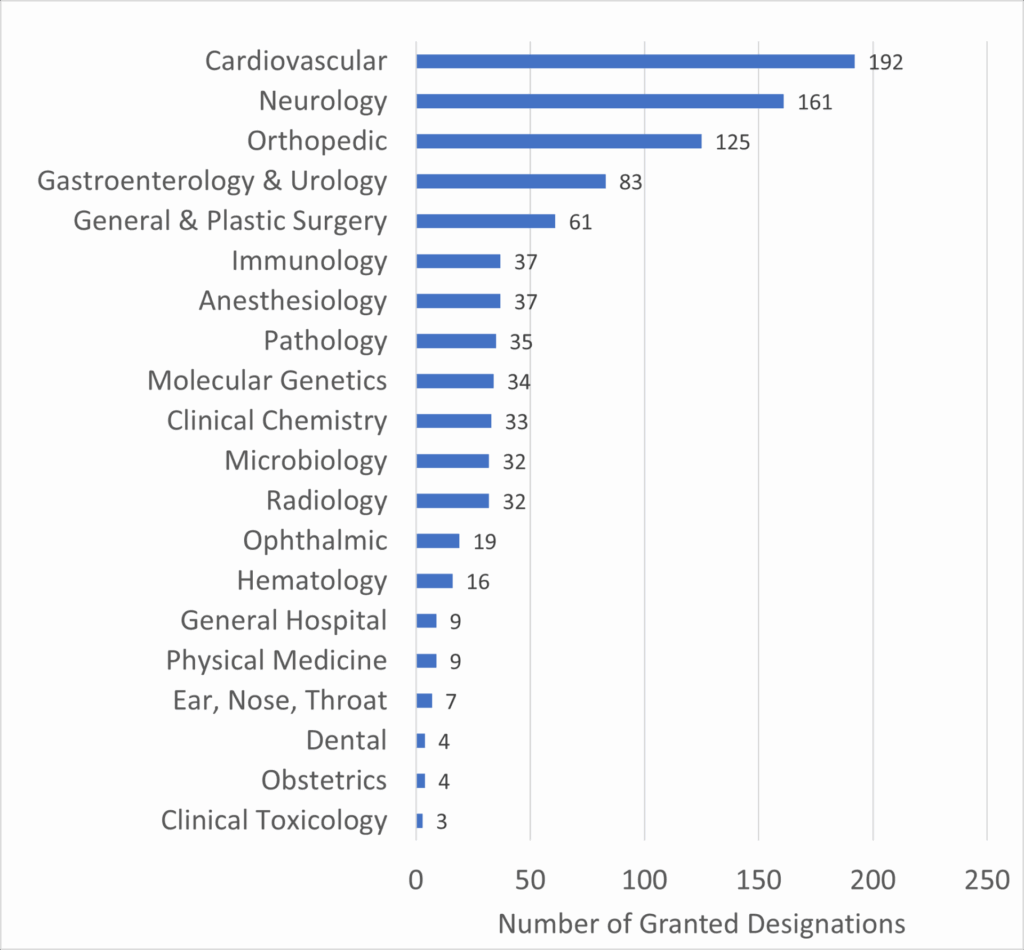
Source: FDA
This data highlights the Cardiovascular and Neurology panels as leading beneficiaries of the Breakthrough Devices Program, which aligns with the high incidence and impact of diseases within these fields. For companies like V-Wave, which operates within the cardiovascular space, the program’s emphasis on these critical areas can significantly enhance the likelihood of successful market entry.
Advantages of the Breakthrough Devices Program
- Accelerated Review Process: One of the program’s most significant benefits is the expedited review process. Devices designated as breakthroughs receive priority review, which significantly reduces the time required for market approval.
- Enhanced Communication: Companies in the program benefit from more frequent and detailed interactions with the FDA. This ongoing dialogue allows for early identification and resolution of potential regulatory issues, helping to prevent delays and streamline the approval process.
- Flexible Clinical Study Designs: The program allows for more adaptable clinical trial designs, which can be particularly beneficial for novel devices that may not fit traditional regulatory pathways. This flexibility enables companies to design studies that are better aligned with their specific devices and patient populations.
- Collaborative Approach: The Breakthrough Devices Program fosters a collaborative relationship between the FDA and device developers. This partnership helps align regulatory expectations with the device’s development, minimizing the risk of unforeseen challenges during the review process.
Challenges and Considerations
While the Breakthrough Devices Program offers significant advantages, it is not without its challenges:
- Stringent Criteria for Designation: Entry into the program is highly competitive. Devices must demonstrate substantial advantages over existing alternatives or provide a more effective treatment for conditions with no approved treatments. Meeting these criteria requires robust evidence and a clear clinical rationale.
- Regulatory Uncertainty: Despite the program’s goal of expediting approval, the pathway is not always straightforward. Companies must navigate the complexities of the FDA’s regulatory requirements, and even with Breakthrough designation, there is no guarantee of approval. This uncertainty can pose risks, particularly for startups with limited resources.
- Resource Intensity: Participation in the Breakthrough Devices Program demands significant resources. Frequent interactions with the FDA, the need for comprehensive clinical evidence, and the iterative nature of the feedback process require considerable time and effort from the development team.
V-Wave’s Strategic Use of the Program
V-Wave’s success in leveraging the Breakthrough Devices Program highlights the potential benefits of a strategic regulatory approach. By securing multiple Breakthrough Device designations for their interatrial shunt, V-Wave was able to capitalize on several key advantages:
- Priority Review: Accelerating the FDA’s evaluation process.
- Enhanced Communication: Engaging in ongoing, productive dialogue with the FDA.
- Flexible Study Design: Tailoring their clinical trials to meet both regulatory and patient needs.
This strategic approach not only facilitated the device’s approval but also made V-Wave an attractive acquisition target for Johnson & Johnson, illustrating the value of a well-executed regulatory strategy.
Conclusion: Lessons for the Medical Device Industry
The FDA’s Breakthrough Devices Program represents a powerful tool for companies developing innovative medical devices. However, success within the program requires a deep understanding of the regulatory landscape, a robust clinical strategy, and the resources to engage effectively with the FDA.
For industry partners, startups, and other stakeholders, V-Wave’s journey through the Breakthrough Devices Program offers valuable insights into how to navigate this complex but rewarding pathway. By studying successful cases like V-Wave, companies can better position themselves to bring groundbreaking technologies to market, ultimately benefiting patients and advancing the field of medical innovation.
References
- U.S. Food and Drug Administration. (n.d.). Breakthrough Devices Program. Retrieved from https://www.fda.gov/medical-devices/how-study-and-market-your-device/breakthrough-devices-program