Does your regulatory strategy indicate that the most appropriate regulatory path to market your device in the US is 510(k)? Did you ask yourself what next?
Consider the following key points:
▪ Start by thinking about what are you going to say about your device when you will market it in the US. ▪ Choose the appropriate predicate device to demonstrate substantial equivalence according to your marketing claims, intended use, and technical characteristics. ▪ Determine the 510(k) types (traditional, special, or abbreviated) and prepare the submission accordingly. ▪ Have all required tests and data to support your submission (using recognized consensus standards and relevant FDA guidance). ▪ Ensure the advocacy of your 510(k), it is not sufficient to have evidentiary documents only. ▪ Ensure the data integrity of your submission and use good submission practices to enable smooth review. ▪ Get to know the eSTAR (Starting October 1, 2023, all 510(k) submissions, unless exempted, must be submitted as electronic submissions using eSTAR). ▪ Understand the impact of MDUFA metrics on FDA actions. ▪ Be familiar with the options to communicate with the FDA (before and during the submission). ▪ Get to know the function of the CDRH Deputy Ombudsman (in case you will need to contact him).
In this post we would like to highlight the relevance of appropriate computer system classification based on GAMP 5 guidelines. GAMP 5 establishes a framework for validating computerized systems in the healthcare sectors. Computer systems should be properly classified in order to determine the amount of risk associated with a system as well as the controls required to reduce those risks. Those actions dictate the validation effort. GAMP 5 divides computer systems into three categories: 3, 4, and 5. (There is another category for software but it’s for Infrastructure Software, Tools, and IT services)
Here is the definition of the different categories:
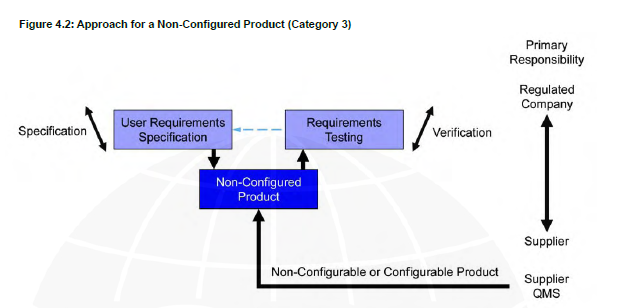
Category 3
Off-the-Shelf products used for business purposes, which includes systems that cannot be configured and that are configurable but for which only the default configuration is used.
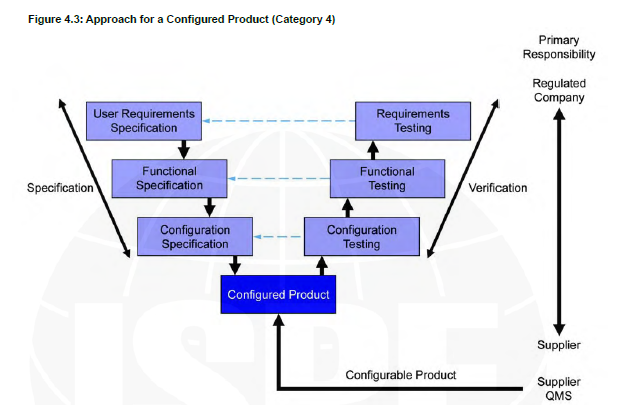
Category 4
Configurable software products that provide standard interfaces and functions that enable configuration of user specific business or manufacturing processes. This typically involves configuring predefined software modules and possibly developing further customized modules.
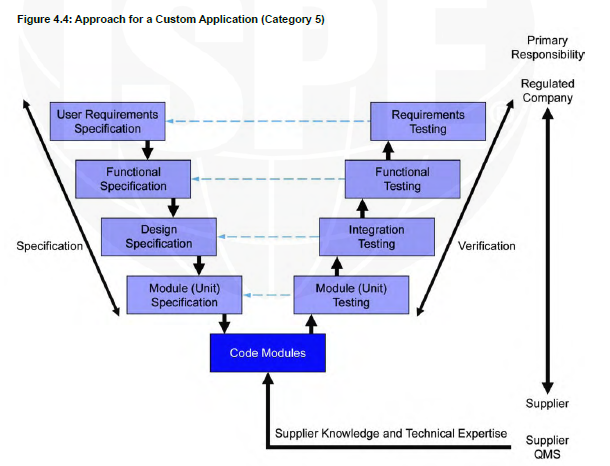
Category 5
Customized systems are developed to meet the specific needs of the users. Custom development may be a completely new system or an extension to an existing system. Complex systems often have layers of software, with one system including components of several software categories; these are commonly treated as Category 5.
V shape model Category 3
For example, UV system that runs on all the spectrumand user’s privileges can’t be changed.
V shape model Category 4
For example, UV system that I can billed different protocol with different light spectrums to fit my needs.
V shape model Category 5
For example, a QMS system that connects to a lab system to collect the date from there and when there is a deviation in the test automatically the QMS will open a deviation form.
As we have seen there is an impact for the different categories on the validation that is need to be performed. So, let’s go to the most important question, How do we determine what category, the system we have or the one we want to purchase? In order to do that we will need first to determine whether this system have any impact on cGxP according to the following bullets:
Automation or control any of: Manufacturing, Sterilization, Formulation, Labeling, Inventory, or Critical Environment Controls
System will be an original source of data for the automation or control of any of: Manufacturing, Sterilization, Formulation, Labeling, Inventory, or Critical Environment Controls
System will use raw and in-process material, clinical data analysis, automated inspection equipment and laboratory data system
System is used to generate, manage and analyze data concerning Product Quality, Safety, Efficacy, Strength Stability or identify
Supporting any GxP Functions such as Calibration, Maintenance Scheduling, and Quality Trending
Manage market complaints or adverse event reporting or electronic document submission/reporting to regulatory agencies
Maintaining copies of protocol pertaining to non-clinical study?
If your answer is No then great, we stop the process here. If you have one answer of Yes, so the system is related to cGxP, and we should move to the next set of questions to decide the systems category.
Was the system developed specifically for the company or any customization done to this application?
If your answer is yes then your system is Category 5 If not we will go to the next question
Is the system a standard product developed by a Vendor where the System-Level Configuration is being modified (excluding Run-Time Configuration) to fit the company’s business process/flow?
If your answer is yes then your system is Category 4 If not we will go to the next question
Is the software a standard product developed by a Vendor and is either
not configurable or
configurable but only the default configuration like run time? (Category 3 – Non-Configured)
If this is the correct then your system is Category 3 If You are not sure contact us for further assistance
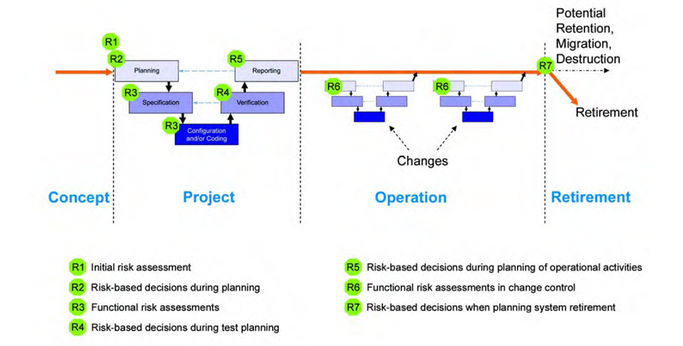
Most companies have a risk methodology in place. In this post, we want to remind you of the critical questions that should guide you during the risk assessment process and the key points where your computerized system can be at risk. But before the questions, do you need to consider your acceptable risk level? The questions we should have in mind: ▪ What can go wrong? ▪ What is the harm? ▪ What is the impact? ▪ What is the probability of a system failure? ▪ What is the detectability of a failure? ▪ How will the risk be managed? The essential thing is to remember that there are risks during the Computerized system life cycle.
If you recognize a risk in one of your systems in one of the different parts of the life cycle, please don’t hesitate to contact us; we would be happy to help you.
The new In Vitro Diagnostic Regulation (IVDR) 2017/746 is a set of regulations issued by the European Union (EU) for the control and supervision of in vitro diagnostic medical devices (IVDs). These regulations aim to improve the safety and effectiveness of IVDs and ensure that they are of high quality.
The IVDR has been applicable since 26 May 2022. In January 2022, the European Parliament and the Council adopted a staggered extension of its transition period, ranging from 26 May 2025 for high-risk in vitro diagnostics to 26 May 2027 for lower risk in vitro diagnostics, and to 26 May 2028 for certain provisions concerning devices manufactured and used in health institutions.
To be prepared for the IVDR, there are several steps that manufacturers, distributors, and other stakeholders can take: Familiarize yourself with the requirements of the IVDR. Make sure you understand the requirements of the IVDR and how they apply to your organization and products. Verify your device classification according to the IVDR new classification system as you may now require the involvement of a Notified Body. Review your current processes and procedures. Ensure that your processes and procedures for developing, manufacturing, and distributing IVDs comply with the requirements of the IVDR. Develop a plan for transitioning to the IVDR, including timelines and budgets. Consider seeking assistance from a third party, such as a regulatory affairs consultancy, to help you navigate the transition to the IVDR. Keep up to date with developments in the IVDR and any changes or updates to the regulations.
By following these steps, you can ensure that your organization is prepared for the IVDR and can continue to provide high-quality IVDs to patients. Contact US for further support and guidance.
This article was prepared by:
Dr. Einat Dekel, DVM
QA & RA Medical Device Senior Consultant, SME In-Vitro Diagnostics & Digital Health
When discussing upgrading the company’s quality system and technical documentation to meet the EU-MDR requirements, many of the companies still think that this is “an activity that QA&RA department should deal with”.
However, the truth is that the main failure cause or bottleneck for upgrading to EU-MDR is the activity related to design and development. For example, product verification and validation tests that have not been performed, design and development documentation that was not kept up-to-date (specifications, drawings etc.), product risk management plans and reports, design changes that were not analyzed and implemented properly and/or were not followed by the relevant verification and/or validation tests, customer complaints that were not analyzed properly as far as influence on product performance and safety is concerned, clinical evaluation documentation including postmarking clinical follow-up and in some cases also the need for clinical trials.
These activities take time, usually, months or even years. Many of these requirements were already there in the MDD, but they were not enforced by the NBs to the extent that they are enforced under the EU-MDR. If the company does not have significant support from the design and development team, the QA&RA departments will not be able to successfully lead the effort to meet the requirements of the EU-MDR. The issues described in relation to design and development, are also critical for successful FDA inspections, which have significantly increased over the last year. It is very important to recognize this fact and understand what needs to be done to achieve compliance.
If you recognize a risk in one of your systems in one of the different parts of the life cycle, please don’t hesitate to contact us; we would be happy to help you.
This article was prepared by:
Marina Lebel B.Sc., CQE
VP, Medical Device
Skip to content window.TEAMME_CONFIG = { companyNamespace: "bringthemhome", };