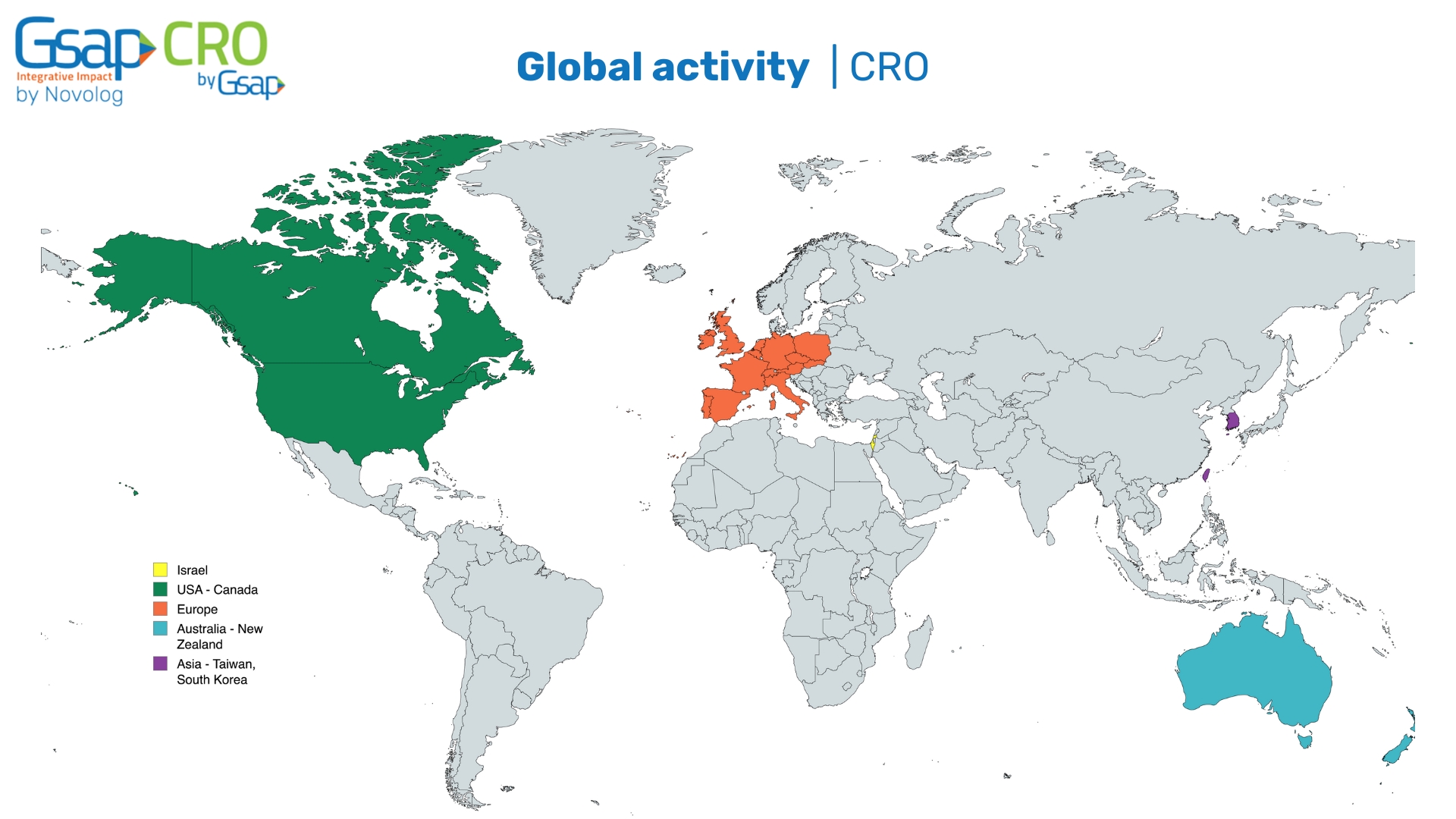
Wound dressings are materials or products applied directly to a wound to promote healing and protect it from infection. They serve several purposes such as protection from external contaminants, absorption of excess wound exudate (fluid), and promotion of healing while providing comfort. Types of wound dressings vary widely and include gauze dressings, foam dressings, transparent films, hydrocolloid dressings, alginate dressings, hydrogel dressings, and others.
Fig. 1. Traditional wound dressings: gauze transparent films, foam dressings, hydrogels, hydrocolloid, and hydro-conductive dressings1.
This review provides highlights of the various regulatory challenges for the approval and clearance of wound dressing products. The path to regulatory approval and data requirements for wound dressing products can vary widely based on many factors, including the product composition, intended use, primary mechanism of action, and treatment indication. Understanding the different regulatory pathways and requirements necessary for authorization along each path is critical to ensure compliance with applicable guidelines and streamline approval processes.
Medical Device or Medicinal Product?
The first challenge is determining whether the product falls under the definition of medical device, medicinal product, or a combination product since a lot of wound dressings are borderline products (those where it is not clear from the outset whether they fall under the MDR or the MPD). This determination can sometimes be complex and depends on the product’s intended mode of action and primary function.
What separates medical device products from medicinal products is the mechanism by which a product exerts its principal intended action. In the case of a medicinal product, the effect is achieved by a pharmacological, immunological, or metabolic mechanism of action, whereas in the case of a medical device, the effective mechanism of action must not be pharmacological, immunological, or metabolic although these mechanisms may assist the device in its function. Medical devices generally achieve their primary intended action through physical, mechanical, or thermal means and they do not typically contain pharmacologically active substances that exert a therapeutic effect like medicines do.
Wound dressings primarily function by providing a physical mode of action that manages the micro-environment of the wound to support the natural healing process and are well accepted as medical devices. For example, the wound dressing may provide a physical barrier, absorb exudate, maintain a moist environment, or protect the wound from external contaminants.
Determining the primary intended action is crucial and is typically described in the manufacturer’s labeling and claims, supported by state-of-the-art scientific data specific to the device. Determination is made on a case-by-case basis and plays a vital role in the device’s classification. Factors taken into account include the device’s intended use, mechanism of action, and how it achieves its therapeutic effect.
It’s important to note that some advanced wound dressings or those containing medicinal substances may require additional regulatory considerations. For example, if wound dressings contain a medicinal ingredient, such as chlorhexidine, where the principal intended action is to provide a local antimicrobial effect, it will be a medicinal product. On the other hand, solutions incorporating substances with ancillary action, e.g. preservatives, remain a medical device.
Some wound dressings may contain both medical device components (e.g., physical structure, absorbent materials) and medicinal product components (e.g., active ingredients, drugs). These are classified as combination products.
Products that straddle the borderline between medical devices and medicinal products may face challenges in classification, as regulations for each category vary significantly in terms of testing requirements, approval processes, and ongoing monitoring. Manufacturers should follow updated guidance documents such as the guidance document released by the Medical Device Coordinating Group to support manufacturers in qualifying borderline products (MDCG 2022-5).Top of Form
Classification of wound dressings
Another challenge involves the classification of wound dressing products based on risk into Class I, IIa, IIb, or III devices. This classification depends on factors such as their intended use, invasiveness, duration of contact with the body, and potential associated risks. For instance, simple wound dressings like non-adherent gauze pads, bandages, and adhesive bandages that do not penetrate the skin are categorized as Class I. Hydrogel dressings designed to manage the micro-environment of superficial wounds that do not breach the dermis are classified as Class IIa, while those intended for wounds that breach the dermis fall under Class IIb.
Verification and Validation of Wound Dressing
Verification and validation (V&V) are critical processes in the development and regulatory approval of wound dressings These processes are planned based on a thorough risk assessment approach. Key potential risks associated with wound dressing devices include:
1. Adverse Tissue Reaction: Ensuring that the materials used in the dressing do not cause allergic reactions or irritation to the patient’s skin or wound site.
2. Delays in Wound Healing: Verifying that the dressing supports and accelerates the wound healing process rather than delaying it.
3. Incompatibilities with Other Therapies: Assessing compatibility with concurrent therapies or medications that may interact adversely with the dressing materials or components.
4. Infection: Testing for the dressing’s ability to prevent microbial contamination and infection at the wound site.
5. Loss of Barrier Function: Ensuring that the dressing maintains its barrier function to protect the wound from external contaminants.
6. Microbial Growth Within the Product: Preventing microbial colonization or growth within the dressing itself, which could lead to infection.
7. Product Degradation During Shelf Storage: Evaluating the stability and durability of the dressing over its shelf life, including factors like temperature sensitivity and packaging integrity.
8. Retention of Dressing Material in Wound: Verifying that the dressing does not leave residues or particles that could impair wound healing or cause discomfort.
These risks guide the specific V&V tests conducted during the development of wound dressings. The selection of test methods is determined by factors such as the type of device, the duration and nature of body contact, and the intended use. Each test aims to mitigate these risks and ensure that the dressing meets the safety, efficacy, and performance standards required for regulatory approval.
For wound dressings, verification involves confirming that the product specifications, materials used, manufacturing processes, and other design elements align with the intended design requirements. This may include testing the physical properties of the dressing, performance, and characterization testing, absorbency, permeability, microbial testing, and biocompatibility.
Validating a wound dressing involves assessing its performance under real or simulated conditions that replicate its clinical use. This includes testing for effectiveness in managing exudate, promoting wound healing, preventing infection, antimicrobial efficacy clinical performance testing, and ensuring patient safety. Validation also considers factors such as shelf-life stability, usability, and compatibility with other medical treatments.
The Changing Regulatory Environment
Another aspect is to constantly be aware of recent updates and guidelines since regulations are continually evolving. Recent updates include the transition from MDD to MDR represents a shift towards more stringent regulation and oversight of wound dressings, to ensure higher levels of patient safety and device performance across the European market. Key changes and implications include stricter requirements and increased emphasis on clinical evidence and post-market follow-up.
Updates in the US include the introduction by the FDA of new proposed rules aimed at classifying previously unclassified wound dressings and liquid wound washes containing antimicrobials. These rules would categorize products based on their level of antimicrobial resistance (AMR) concern. Products with a high level of AMR concern would fall under class III medical devices, necessitating Premarket Approvals (PMAs), while those with medium or low levels of concern would be classified as class II devices. These proposed rules, when become effective, will impact both existing commercially available devices in this category and future products.
Manufacturers of wound care products incorporating antimicrobials or other chemicals should analyze their products to determine if they could be affected by these proposed rules. They should also assess their regulatory status under the new classification to ensure that their current marketing authorization remains adequate. Additionally, companies may need to update their product labeling and review their marketing claims to align with the new regulatory requirements, as failure to do so could result in their products being reclassified as combination products or drugs.
In summary, different regulatory pathways, with varying degrees of data and regulatory oversight, can be used to achieve market authorization for wound dressing products. Navigating these regulatory requirements requires a thorough understanding and adherence to applicable guidelines to ensure the safety and efficacy of wound and skin care before placing products onto the market. In addition, the specific regulations may vary depending on the country or region and it is recommended to consult with regulatory authorities or seek professional advice to ensure compliance with specific regulations in the relevant country or region.
At Gsap, we collaborate with our in-house medical device experts, pharmaceutical experts, clinicians, and technical teams to cover all areas of medical devices including medical devices used for wound and skin care, and confirm the product’s readiness for market.