

RNA therapeutics have finally arrived and taken their place as a viable drug discovery platform [1]. Their potential to increase, by orders of magnitude, the number of druggable targets, was evident from the first FDA approval of an RNA therapeutic back in 1998. Only now, however, is this potential being realized with 28 RNA therapeutics now approved globally and in terms of the number of products in development, RNA therapeutics are overtaking unmodified cell therapies [2].
The emergency use and approval of the two mRNA-based vaccines from BioNTech and Moderna shone a spotlight on the potential of RNA-based medicines as a whole, and the speed with which they were manufactured to combat the COVID pandemic gave research towards RNA therapeutics development an immense, and much needed, boost.
In contrast to small molecule drugs and larger biologics, high-quality RNA constructs can be generated faster, and at lower costs; their manufacturing process platform can support any RNA sequence, allowing for personalized RNA therapeutics; and since RNA doesn’t integrate into the host genome, RNA therapeutics have an improved risk/benefit profile.
RNA’s intermediary position in the expression of genetic information from DNA to protein presents a huge number of pharmacological targets that were previously undruggable by monoclonal antibodies or small molecules. Moreover, this central position provides a unique versatility to modulate gene expression to introduce new transcripts for protein replacement therapy and more [3]. RNA therapeutics are a diverse group and span from antisense oligonucleotides (ASOs), small interfering RNA (siRNA), microRNA (miRNA), and messenger (mRNA). In general, ASOs and RNA inhibition (RNAi) therapeutics promote RNA degradation and inhibit translation, whereas mRNA therapeutics promote protein or antigen expression. Based on a recent survey of the RNA-based therapy landscape [4], products leveraging RNAi and mRNAs make up the largest portion of the pipeline at 40% and 37% respectively, but the broader pipeline includes oligonucleotide, double-stranded RNA (dsRNA), and micro-RNA (miRNA) products as well.
RNA, in contrast to DNA, is remarkably unstable and is rapidly degraded by RNases which are ubiquitous in the environment; RNAs’ often large size and strong negative charge hinder transport across the cytoplasmic membrane; and exogenous RNA can be highly immunogenic, promoting cell toxicity and impairment of translation into therapeutic proteins. The recent rapid growth of RNA therapeutics has been due to successes in addressing these challenges of stability, delivery, and immunogenicity; including the ability to penetrate the cell membrane and an ability to escape endosomal entrapment once inside the cell. Chemical modifications of RNA facilitated the shift from completely encapsulated RNA nanoparticles to the use of less complex RNA conjugates (e.g., GalNAc). More recent approaches include circular RNA which is stable against exonucleolytic decay, and bioengineered RNA agents produced and folded in living cells also indicate a favorable stability in human cells [5, 6].
Improvement and innovations have accelerated now that RNA Therapeutics are unequivocally feasible, but drug candidates yet need to complete many steps before clinical use, including manufacturing according to Good Manufacturing Practice (GMP) guidelines, pharmacokinetic (PK) / pharmacodynamic (PD) studies, and safety evaluations. Hospital-based RNA therapeutics programs were predicted to be at the forefront of RNA-based drug development, being best positioned to accelerate the translation of transformative therapies from the lab bench to the patient’s bedside [5].
In the US, some RNA-based therapies are regulated as gene therapies, such as those with viral vector delivery systems and mRNA vaccines; these are regulated as biological drugs under a Biologics License Application (BLA). On the other hand, other RNA-based therapies, such as RNAi products are regulated as small molecule drugs under a New Drug Application (NDA) [4]. It’s important to note that in the US these pathways differ in terms of the lengths of marketing exclusivity awarded upon authorization, and the barriers to competition are different for generic compared to biosimilar products.
Gsap’s Advanced Therapies group makes it their business to stay on top of the latest developments in RNA Therapeutics and intends to leverage our extensive regulatory, manufacturing, and development expertise with Advanced Therapies to help clients develop their novel products for marketing approval. Gsap has considerable experience working with the major Israeli hospitals in establishing cGMP-compliant manufacturing capabilities and quality control methods. We’re familiar with navigating unchartered territory. We’re ready and looking forward to guiding clients along a regulatory-compliant critical development path to market.
Figure 1: Various RNA Therapeutics
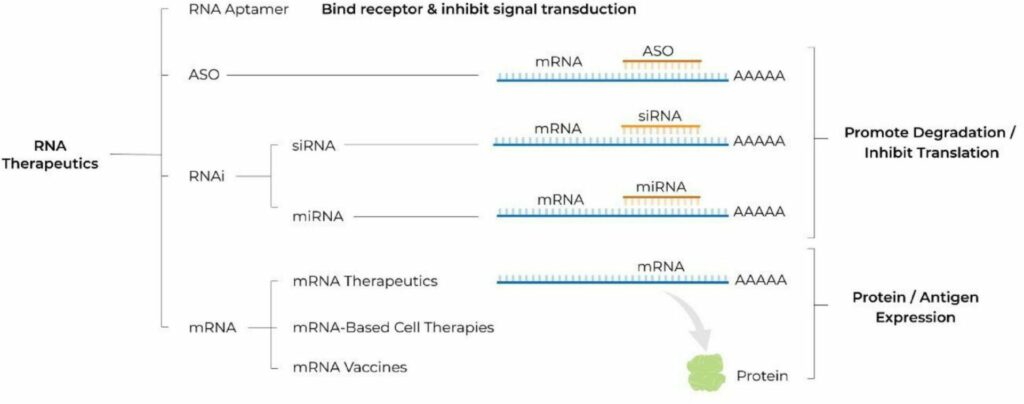
From Damase et al., The Limitless Future of RNA Therapeutics. Front Bioeng Biotechnol. 2021 Mar 18;9:628137. doi: 10.3389/fbioe.2021.628137. Copyright © 2021 Damase, Sukhovershin, Boada, Taraballi, Pettigrew and Cooke.
References:
- Agrawal S. RNA Therapeutics Are Stepping Out of the Maze. Trends Mol Med. 2020 Dec;26(12):1061-1064. doi: 10.1016/j.molmed.2020.08.007. Epub 2020 Sep 25.
- Data source: Gene, Cell, + RNA Therapy Landscape Report, American Society of Gene & Cell Therapy and Citeline, 2024. https://www.asgct.org/publications/landscape-report
- DeWeerdt S. RNA therapies explained. Nature 574, S2-S3 (2019) https://doi.org/10.1038/d41586-019-03068-4
- Overview and Outlook for RNA-Based Therapies – White Paper. Avalere Health (2024) https://avalere.com/wp-content/uploads/2024/06/20240522-Lilly-RNA-Based-Therapies-White-Paper-vFINAL.pdf
- Damase TR, Sukhovershin R, Boada C, Taraballi F, Pettigrew RI, Cooke JP. The Limitless Future of RNA Therapeutics. Front Bioeng Biotechnol. 2021 Mar 18;9:628137. doi: 10.3389/fbioe.2021.628137.
- Dammes N, Peer D. Paving the Road for RNA Therapeutics. Trends Pharmacol Sci. 2020 Oct;41(10):755-775. doi: 10.1016/j.tips.2020.08.004. Epub 2020 Sep 3.















{kind=link}