Achieving CE marking for a medical device starts with one of the most critical components in the EU regulatory process: the Technical File. But navigating the requirements of the EU MDR—and aligning your documentation with the expectations of Notified Bodies—can be complex.
EU MDR Technical File for Medical Device Submissions Webinar
In this expert-led webinar, Gsap and Dekra join forces to walk you through the essential elements of an EU MDR-compliant Technical File. From structure to strategy, the session provides practical guidance for regulatory professionals, QA teams, and innovators preparing for EU market access.
🎯 What You’ll Learn:
A step-by-step breakdown of the EU MDR Technical File structure
Common mistakes in submissions—and how to avoid them
What Notified Bodies really look for during document review
How to align your file with MDR Annex II & Annex III
Tips for building audit-ready, CE-markable documentation
Whether you’re preparing your first submission or refining an existing one, this session equips you with the tools to streamline your process and increase your chances of successful EU approval.
Clinical trials are at the heart of medical innovation—but they’re not without challenges. From rising costs and recruitment hurdles to regulatory complexities, sponsors and CROs are constantly looking for ways to conduct trials more efficiently and effectively. This recorded webinar brings together experienced voices from Gsap and Medistat to share practical insights for optimizing clinical trial operations, particularly when working with limited resources or under tight timelines.
🎯 What You’ll Learn:
How to reduce trial costs and improve operational workflows
Strategies to enhance patient recruitment and retention
Regulatory insights to avoid common delays and pitfalls
Smarter budgeting techniques for early‑stage and growing companies
Real‑world case examples from both CRO and sponsor perspectives
Whether you’re leading a clinical team, supporting trial execution, or managing strategy and compliance, this session offers valuable takeaways to help you move forward with greater clarity and control.
Packaging validation is a critical step in ensuring product safety, sterility, and compliance with global regulatory requirements.
In this recorded webinar, experts from GSAP and Medimor Ltd. explore the practical aspects of building a compliant packaging system and executing validation processes that meet international standards such as ISO 11607, FDA, and EU MDR.
🎯 What You’ll Learn:
How to design and validate a compliant sterile barrier system
Overview of key validation tests: ASTM F1886, F1929, F1140, F88, and more
Common regulatory pitfalls and how to avoid them
Real-life examples from validated packaging systems
Discussion and Q&A with validation professionals
Whether you’re involved in QA/RA, validation, production, or regulatory strategy, this session will help you ensure packaging integrity throughout the product life cycle.
Bringing a digital health product to market is complex, but with the right guidance, it’s achievable.
In this expert-led webinar, professionals from Gsap and Matrix Medika share practical strategies to help you navigate the key challenges in regulation, quality, and development for digital health products in the EU and U.S.
🔍 What you’ll learn:
How to streamline your product development process
How to avoid common regulatory and quality pitfalls
How to reduce delays and improve submission success
How to approach FDA and MDR approvals more confidently
Designed for startups, developers, and QA/RA professionals, this session is especially relevant for those working with SaMD, AI-based tools, and other digital health technologies.
At Gsap, we take pride in providing end-to-end support across quality, regulation, and clinical trial management (Gsap CRO), ensuring our clients achieve their goals efficiently and successfully.
Gsap CRO planned, executed, and managed the study end-to-end until the final CIR.
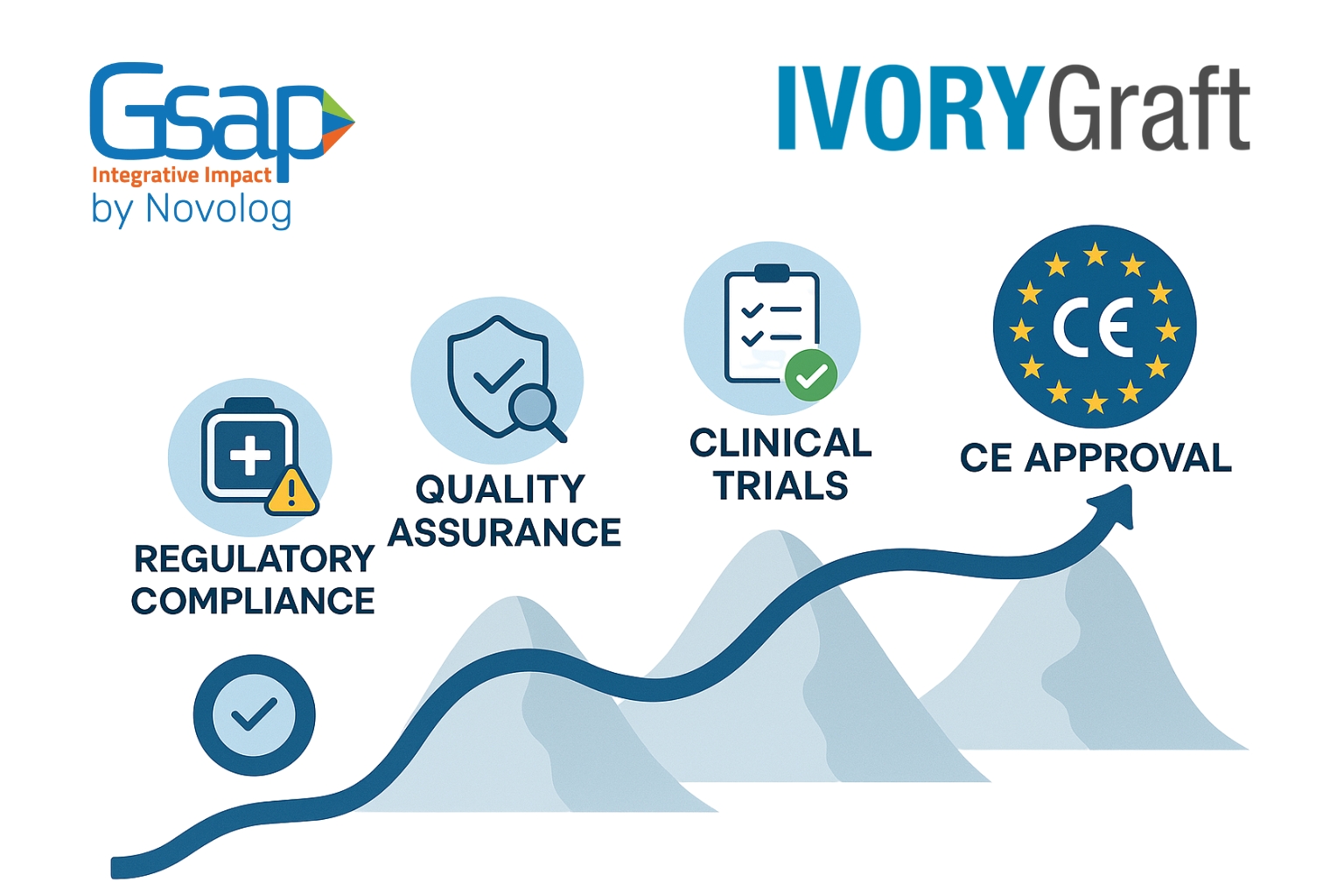
This month, we are thrilled to mark the completion of a long-term clinical study for Ivory graft, which developed Ivory Dentin Graft, for which we have participated for the past eight years.
Throughout this time, we managed the clinical trial, solved regulatory questions, and quality processes, leading to a significant milestone—CE approval for their Class III medical device!
This achievement is a testament to the multi-disciplinary impact Gsap brings to the industry. By integrating expertise across multiple domains, we accelerate innovation, reduce regulatory hurdles, and drive successful market entry for cutting-edge healthcare solutions.
This month, we are excited to announce the successful completion of the clinical study, which included the completion of 30 patients and obtained valuable insights.
Ivory Graft designed and developed the first porcine dentin xenograft. Ivory Dentin Graft™ is a dentin bone graft that retains the natural dentin form as well as the natural protein matrix and collagen, important for bone regeneration.
According to Amir, Ivory Graft CEO, “The Ivory Graft’s clinical results show superiority over porcine xenograft material, which is used in the dental industry today, due to the nature of dentin and its positive effect on bone growth.”
Partner with Gsap and experience the power of true end-to-end support. Our integrative approach—spanning clinical trial management, regulatory guidance, and quality assurance—ensures every milestone is met with precision and expertise. Let us help you accelerate innovation, overcome regulatory hurdles, and achieve outstanding execution for lasting impact in the healthcare industry.
Artificial Intelligence (AI) is revolutionizing healthcare, offering unprecedented advancements in diagnostic accuracy, personalized treatment plans, and operational efficiencies. From AI-driven imaging analysis to predictive analytics, these technologies are becoming integral to modern healthcare systems. Reflecting this rapid growth, as of September 2024, the U.S. Food and Drug Administration (FDA) has authorized over 985 AI/ML-enabled medical devices, signifying a substantial increase from just a few dozen devices years prior (Figure 1). The global AI in healthcare market is projected to reach $194.4 billion by 2030, growing at a CAGR of 38.4% from 2022 to 2030.
Figure 1.Graph depicting the evolution of approval numbers for various medical panels over the years, highlighting the dynamic growth and potential research and development focuses in these areas.Credit: Joshi G, Jain A, Araveeti SR, Adhikari S, Garg H, Bhandari M. FDA-Approved Artificial Intelligence and Machine Learning (AI/ML)-Enabled Medical Devices: An Updated Landscape.
However, AI integration presents challenges, including recalls. A 2023 study found 211 AI/ML medical device recalls between 2019 and 2021, mostly moderate risk. Examples include software errors in radiation therapy (K190387) and cardiac ultrasound (K20062). Managing these risks is crucial for AI’s future in healthcare.
Each region has its definitions and guidelines governing AI in healthcare, making compliance a critical yet intricate task for innovators. This article delves into how regulatory authorities define AI, explores global regulatory frameworks, and offers insights on successfully bringing AI innovations to market.
Product/Technology Definition
Understanding how regulatory bodies define AI is crucial for compliance and successful market entry. Below are specific definitions from key regulatory authorities:
United States (FDA):
Artificial Intelligence is a machine-based system that can, for a given set of human-defined objectives, make predictions, recommendations, or decisions influencing real or virtual environments. Artificial intelligence systems use machine- and human-based inputs to perceive real and virtual environments; abstract such perceptions into models through analysis in an automated manner; and use model inference to formulate options for information or action.
European Union (EU):
Artificial intelligence (AI) refers to systems that display intelligent behavior by analyzing their environment and taking actions – with some degree of autonomy – to achieve specific goals. AI-based systems can be purely software-based, acting in the virtual world (e.g., voice assistants, image analysis software, search engines, speech, and face recognition systems) or AI can be embedded in hardware devices (e.g., advanced robots, autonomous cars, drones, or Internet of Things applications).
Key AI Technologies in Healthcare:
Machine Learning is a set of techniques that can be used to train AI algorithms to improve performance at a task based on data.
Natural Language Processing (NLP): Enables computers to understand and interpret human language, facilitating the analysis of clinical documentation.
Computer Vision: Allows machines to interpret visual data from medical images like X-rays, MRIs, and CT scans.
Figure 2. Artificial Intelligence (AI) is an interdisciplinary area demonstrated by machines operating human tasks such as speech recognition,
Global Regulatory Frameworks for AI in Healthcare
Comparison of Global Regulatory Frameworks
Navigating the complex regulatory environment requires an understanding of how different regions approach AI in healthcare.
In the U.S., AI healthcare products are regulated by the FDA under existing medical device frameworks. For approval, your product must go through one of three main premarket pathways: 510(k), De Novo, or PMA (Pre-Market Approval). The 510(k) premarket notification is for products that are similar to an existing, already-approved device, allowing for a quicker clearance process. If your product is new, medium risk, and does not have a comparable product on the market, you will likely go through the De Novo process. For high-risk AI products, you will need to submit a premarket approval (PMA), which requires a comprehensive data set to prove the product’s safety and effectiveness.
In addition to premarket submissions, the FDA emphasizes Good Machine Learning Practices (GMLP) to ensure that your AI system follows best practices in data management, transparency, and reliability. Those practices are implemented in a Total Product Lifecycle (TPLC) approach. This means that even after your AI product is on the market, you must continuously monitor its performance, update it if necessary, and report any issues to ensure ongoing safety and effectiveness.
In the European Union, AI products used in healthcare are regulated under the Medical Device Regulation (MDR), which includes a thorough process to assess product safety and effectiveness. One of the first steps is risk classification, where you determine how risky your AI product is to patients or users. Higher-risk products undergo stricter review. After classification, the next step is the conformity assessment, where you work with a “Notified Body” (an official EU organization) to prove that your product meets the required safety and performance standards. In addition, the EU AI Act is a comprehensive regulation that will impose requirements on AI systems, particularly those deemed high-risk, across various sectors including healthcare. It complements existing regulations like the Medical Devices Regulation (MDR) and In Vitro Diagnostic Regulation (IVDR). For healthcare AI, the Act focuses on transparency, accountability, and risk management throughout the AI lifecycle. High-risk AI systems in healthcare, such as those used for emergency triage or determining eligibility for health services, will face stringent requirements. The Act aims to ensure patient safety and rights while fostering innovation in AI-enabled healthcare technologies.
Both the FDA and EMA frameworks require not only upfront proof that your AI product works but also ongoing efforts to ensure safety and effectiveness even after the product is on the market.
Case Study: FDA Approval of Viz.ai
Background
Viz.ai is an Israeli company founded in 2016 pioneering the use of artificial intelligence to accelerate the detection and treatment of strokes. The company’s AI-powered platform is designed to analyze medical imaging and automatically detect large vessel occlusion (LVO) strokes, potentially life-threatening conditions that require immediate treatment. The platform alerts stroke teams directly through their mobile devices, significantly reducing the time to intervention, which is crucial in minimizing the damage caused by strokes.
Regulatory Pathway
As can be seen in Table 2, Viz has received multiple device clearances from the FDA. Starting with the DeNovo reclassification that created new product codes, and then following with 510K submissions on modifications or new features. Viz shows us that developing a regulatory plan in parallel with your development plan is critical to releasing products to the market in an efficient manner. Note that the first clearance was only 2 years after the company was established.
A key aspect of Viz.ai’s regulatory strategy is its focus on leveraging programs that expedite market entry. For instance, Viz.ai received the FDA’s Breakthrough Device Designation, which fast-tracks the review process for technologies that provide more effective treatment or diagnosis of life-threatening conditions. Additionally, the DeNovo approval for ContaCT in 2018 (DEN170073) allowed Viz.ai to introduce a tool that identifies and communicates specific patient images to specialists, supporting decision-making in a parallel workflow without disrupting standard care.
Viz.ai supported this with a retrospective study to assess the sensitivity and specificity of ContaCT’s image analysis and notification system, using 300 CT angiogram studies from two U.S. clinical sites for comparison against neuro-radiologist assessments.
This early achievement paved the way for ongoing improvements and feature expansions through subsequent 510(k) submissions, as outlined in the table above. These regulatory advancements allowed Viz.ai to continually refine and enhance its technology, ensuring it remains at the forefront of innovation in patient care and diagnostic efficiency.
Compliance with FDA Definitions and Requirements
Alignment with FDA’s AI Definition: The Viz.ai platform aligns with the FDA’s definition of AI-based medical devices. It uses AI algorithms to assist in the real-time analysis of CT scans and other medical imaging, mimicking cognitive functions related to human intelligence in the decision-making process.
Safety & Efficacy: Viz.ai provided extensive clinical performance data showing a reduction in time from image acquisition to stroke team notification by 52 minutes, significantly improving patient outcomes.
This real-time analysis and alert system help reduce the time to treatment, which is crucial as “time is brain” in stroke management.
Outcome
FDA Clearance: Viz.ai’s LVO Stroke Platform became the first FDA-approved AI platform for stroke detection. The platform is now used in over 1,000 hospitals across the U.S., Israel, and Europe.
Impact: By reducing the time to treatment, Viz.ai has significantly improved stroke care, providing earlier intervention, which leads to better patient outcomes and reduced long-term disability rates. The AI-powered alert system ensures that stroke teams are notified promptly, saving critical time in decision-making and patient care.
Lessons Learned
Innovation and Collaboration: Viz.ai’s success shows how combining innovative AI technologies with ongoing and early regulatory engagement can bring transformative healthcare solutions to market quickly.
Proactive Regulatory Approach: By engaging with the FDA’s Breakthrough Devices Program early, Viz.ai was able to expedite the regulatory process, ensuring faster market approval.
Real-World Impact: The success of Viz.ai demonstrates that AI can revolutionize the speed and effectiveness of treatments for time-critical conditions, such as stroke, ultimately saving lives.
Conclusion
The integration of AI into healthcare is revolutionizing the field, offering groundbreaking advancements in diagnostics, treatment, and operational efficiency. However, navigating the complex global regulatory landscape is essential for these technologies to succeed. Different regulatory bodies, such as the FDA and EMA, provide specific frameworks that innovators must align with, ensuring safety, effectiveness, and compliance throughout the product lifecycle. Early regulatory engagement, as shown in case studies, can expedite approval processes, while ongoing compliance with data privacy and safety standards remains crucial. Companies that prioritize regulatory strategy are better positioned to bring innovations to market swiftly and safely, driving progress in this transformative era of healthcare.
Stay tuned for upcoming editions of ‘Innovation Meets Regulation,’ where we explore the intersection of healthcare innovation and regulatory frameworks that shape the industry’s future.
Innovation Meets Regulation – How medical regulations shape the development and success of innovative healthcare products
Bringing a new drug or biologic to U.S market is a complex journey that requires strict compliance with the FDA’s regulations. One key aspect of this process is the Bioresearch Monitoring (BIMO) program, which assures the quality and integrity of clinical trial data.
This article provides an overview of the guidance, its goals, and how it impacts companies preparing submissions. Whether you’re new to regulatory processes or an experienced professional, this guide will clarify what you need to know and do.
What is the FDA BIMO Guidance and Inspection Process?
The Bioresearch Monitoring (BIMO) program is a comprehensive FDA initiative designed to assure that clinical trials are conducted ethically and that their data is trustworthy. Here’s what it entails:
Data Integrity: The program verifies that clinical trial data submitted to the FDA is reliable and accurately reflects study results.
Ethical Compliance Assurance: It ensures that trials comply with Good Clinical Practices (GCP) and that participants’ rights and safety are prioritized. This sustained process helps maintain trust in trial conduct.
BIMO Inspections: The FDA conducts on-site inspections, data audits, and remote regulatory assessments of clinical trial sites, sponsors, contract research organizations (CROs), and institutional review boards (IRBs) involved in the studies. These inspections often follow a risk-based approach to target areas of concern.
BIMO findings provide valuable information to the FDA regarding the integrity and reliability of the clinical data submitted in NDAs and BLAs. This ensures that the data is adequate to support the approval of new treatments. Without this comprehensive program, patient safety and public trust in the drug approval process could be at risk.
The New Guidance for BIMO Inspections: Aim and Main Changes
Aim of the FDA BIMO Guidance
The new guidance aims to standardize and simplify data submission required for planning BIMO inspections. This standardization facilitates the FDA’s ability to identify sites for inspection, evaluate study integrity, and maintain an efficient review timeline.
Standard Data to Submit as Part of NDAs and BLAs
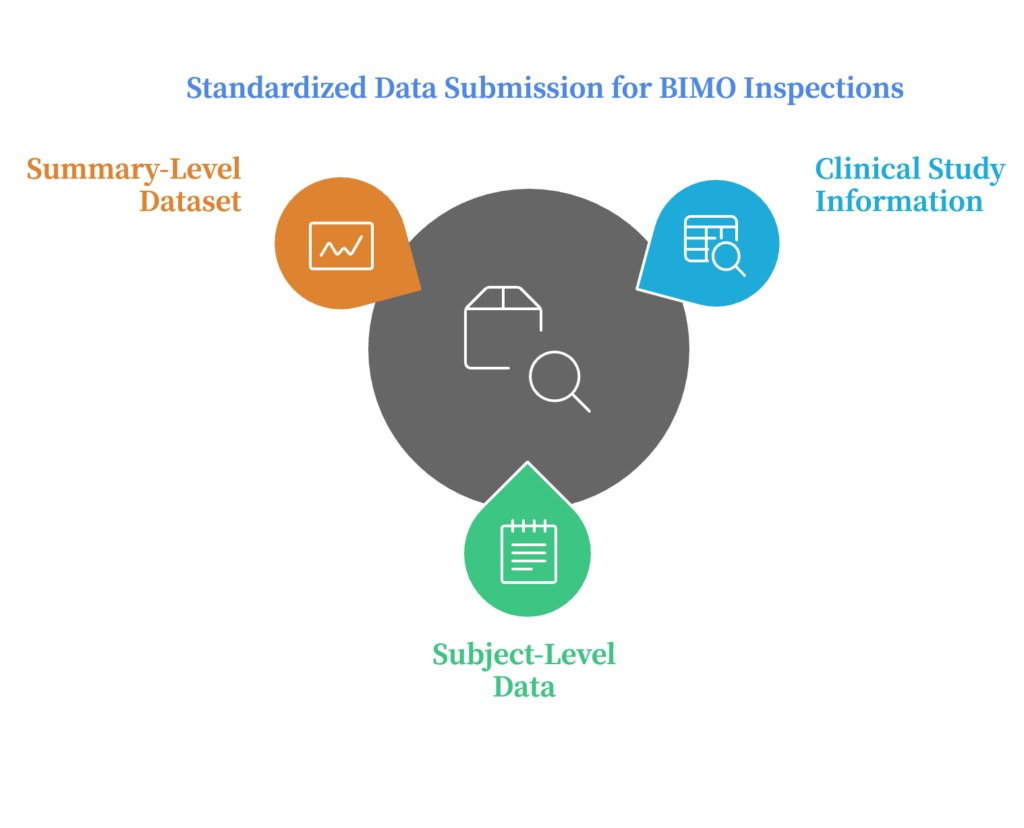
Study-specific data (both clinical study-level information and clinical site data) should be submitted at several levels:
Clinical Study-Level Information: Sponsors must provide comprehensive tables listing all clinical sites, including:
a. Investigator names, and complete site details
b. Details of CROs/vendors or other entities to whom the Sponsor delegated activities.
c. Protocol (and amendments) and Annotated CRF
Subject-Level Data Line Listings: Detailed, site-specific data for each trial participant must be provided, including studies with different treatment indications. Primary data points (in addition to derived data) must be submitted. For example, in a pain trial, raw diary entries of participants’ pain scores (primary data) must be submitted alongside calculated results (derived data).
Summary-Level Clinical Site Dataset: This dataset summarizes key information from clinical sites across all pivotal studies. It allows the FDA to effectively prioritize sites for inspection.
All submissions must adhere to the FDA’s electronic common technical document (eCTD) format and be uploaded through the FDA’s secure gateway.
Key Components of Data Standardization of Submission Content – FDA BIMO Inspection Guidance
What Does This Mean for Your Business?
These changes streamline regulatory submissions and improve the efficiency of FDA reviews. However, they also require more detailed preparation and coordination. Sponsors should consider the following actionable steps:
Streamlined Submissions: Ensure data formats are standardized to reduce variability and errors. This will enable smoother FDA reviews and minimize back-and-forth requests for clarification.
Risk-Based Inspections: Be aware that the FDA employs a risk-based model to prioritize site inspections, meaning sites with incomplete or inconsistent data may face closer scrutiny. Proactively address potential gaps in your data.
Early Integration: Incorporate the guidance’s requirements into clinical trial planning from the outset to avoid delays during submission. Early compliance ensures smoother regulatory processes.
Steps to Stay Ahead
Prepare for Raw Data Requirements: Configure your EDC system to capture detailed raw data, not just calculated scores. Implement edit checks to verify data accuracy and plan for source data verification (SDV) for critical measurements.
Enhance Risk Management: Assess how these requirements may impact your risk management processes and address vulnerabilities early.
Foster Targeted Collaboration: Align regulatory, clinical, and data management teams to address the specific demands of the new requirements. Early coordination is key to streamlining processes and avoiding delays.
Enhance Team Readiness: Sensitize stakeholders, including investigators, CROs, and vendors, about the requirements and their role in providing data to the FDA. Enhanced scrutiny may increase the likelihood of regulatory inspections.
Conclusion
The FDA’s updated BIMO guidance and inspection processes reflect its efforts to prioritize efficiency in regulatory processes while enabling enhanced scrutiny of sponsors, investigators, and CROs. By adopting the standardized format for data submission, companies can expedite their review timelines and demonstrate a commitment to transparency and data integrity.
With the IVDR entering into force on 26 May 2022 in Europe, manufacturers have switched from Europe to the US market as their primary entrance market. To assist the manufacturers, Gsap has joined forces with MDC Associates, a leading US company in the field of IVD, to bring the most updated information to ease navigation through the changing regulatory path.
Join this insightful webinar, led by Gsap and MDC experts, to hear about the latest IVD regulatory updates in the US and their implications for Israeli manufacturers.
Process validation in the medical device industry plays a crucial role in ensuring a successful and effective manufacturing flow. The various challenges associated with process validation can be demanding and often require innovative, out-of-the-box solutions.
Gsap, in collaboration with Flex, has joined forces to address the most common challenges in the process validation (PV) process. To contribute to the successful completion of PV, we hope to impart to our community our vast professional knowledge and practical expertise.