
Digital health revolution in Israel- accelerates by Gsap


Gsap is excited to invite you to the MDI Expo 2021 venue-Where the whole Israeli medical devices community meets!
MDI Expo 2021 convention will be held on 09.11.2021 at the Avenue Congress Center – Airport city.
Gsap experts will provide professional lectures:
“Process Validation in Medical Device” by Haim Eliyahu, Engineering and Validation Project Manager
“Digital Health- The way you never looked at it” by Dr. Ella Sheiman, RA/QA Project Manager
Join us for theMDI Expo 2021:
https://app.icount.co.il/m/e583e/c7924p1u61305fb2274

So, you want to validate your production processes. This is a great start!
There are several benefits to it as shown in this article, but…. where to begin with? How to do it? To which extent is the Validation necessary? How many samples do I need to show compliance? Not to worry, these are common questions regardless of the size of your company or your level of experience.
This article describes how to succeed in Process Validation, with real-life examples of recent Gsap projects, including the following subjects:
In the past, most manufacturers relied on the basic IQ-OQ-PQ protocols but with time regulations became more sophisticated. The good news is that for some cases the regulatory issues can even reduce the Validation burden. Here’s when Gsap will be most helpful for your company and your project.
Let’s begin with the FDA 21 CFR Part 820.75(a), which exempts you from Validation if you have a fully verified process. Yet, how to identify to which extend your manufacturing process is under 100% scrutiny? Is it “fully” verifiable?
Similarly, ISO 13845 releases your process from being validated where the outputs are subsequently monitored and measured. In this case, the question becomes how precise and reliable is your control over the entire process or sub-processes?
The Process Validation Guidance GHTF/SG3/ N99-10, in turn, has provided us with a helpful decision tree. It releases the manufacturer from Validation when verification alone eliminates unacceptable risks. But is verification a cost-effective solution? Again, the Validation question remains.
In practice, the Medical Device industry is generally composed of very complex manufacturing processes, sometimes involving a set of multi-part sub-processes out-sourced in many instances. So, to what extent should the Validation take in-house and outside? Still, regardless of the regulatory issues, is your customer requiring validations? What then should be the actual scope of work?
This is when you need a professional to help to decide which is the best course of action Validation will take for your business.

Gsap has experienced professionals who are aware of the different companies and their sizes, manufacturing volumes, organizational structures, and management styles. In this newsletter, we illustrate successful projects which were validated to ensure that processes operate within specifications and consistently produce safe products, all of them in compliance with the quality predetermined requirements.
One of our world-class clients has complex assembly lines for different Class II and III products. The FDA has required a confirmatory validation for all platforms. The goal was clear. Our professionals learned the processes in-depth, mapped the manufacturing flows, and developed the IQ-OQ-PQ protocols including Test Method Validation for specific continuous and discrete parameters. Five consecutive runs for each platform were analyzed, recorded and the results proved that each production line can consistently satisfy the requirements. That is to say, it was confirmed that all Device Master and History Records (DMR’s and DHR’s) were suitable for use in production. The project goal was achieved in time within our dedicated-for-client methodology and compliance.
The cornerstone preceding a successful Process Validation is the documented Risk Management file. Based on it, the Process Validation Plan indicates the actions needed to ensure that production risks, including software risks, are under control and do not violate product specifications. However, there are cases when mitigations adopted do not sufficiently cover all safety concerns for the product. As a result, residual risks become unacceptable.
We at Gsap work with what most manufacturers use for this preliminary phase: The pFMEA technique (process Failure Mode & Effect Analysis). There are three basic questions to be answered: Starting with “What can go wrong?”, we then move to “What are the chances it could go wrong?”, and finalize by asking ” If it goes wrong what are the consequences?”. The mitigations and control possibilities shall follow the assessment.
Professional support here may not only analyze and develop the Risk Management Process but also distinguish between the pFMEA and dFMEA (design FMEA). In short, Process Validation shall be effectively focused on the parameters that reflect the risks involved in your manufacturing process.

A committed Validation team is what makes the project flow.
One thing to keep in mind is to form a validation team even before the project begins. Field experience has shown that managers who gave this step the right importance had their projects flowing at a fast pace. Defining responsibilities and required deliverables from the different players is also key to the success of a Process Validation Project.
The Validation team should have representatives from at least Engineering, Quality Assurance, and Operations. Laboratories, R&D, Regulations, and Purchasing are recruited on a case-by-case basis.
The main roles of the team are:
●Early identification of the existing processes and sub-processes
●Detecting existing and potential monitoring and control features
●Approvals of the Process Validation Plan (PVP), Protocols, and Reports.
Some manufacturers call the PVP the Master Validation Plan, a document that describes the purpose, the scope and approach to be taken for the validation work, a description of the manufacturing processes, the Validation phases and associated requirements, the scheduled of work, and foreseen cases for revalidations. The MVP shall refer to relevant technical documentation, Work instructions, Sampling, and Criteria for success. In some cases, it also includes a list of tools involved in the processes.

This is the core of Process Validation. Generally being time and work-consuming, the qualifications start with one question as well put by the PV guidance: “Is the equipment properly installed?”. This is the Installation Qualification (IQ) phase of the project. The focus turns to the equipment design, installation features, cleaning requirements, environmental conditions, calibrations, safety, software, and technical documentation. This is also the phase when equipment functionality is put to prove generally off-line.
Once the infrastructure, the environmental conditions, the mechanical, electrical, and similar requirements are fulfilled, documented, and acceptable to run the process, the Installation Qualification phase is formally approved and considered finished.
Next, the attention is driven to the operational ranges which allow manufacturers to produce products inside the specifications. This is the Operational Qualification (OQ) phase. At first, a review of all process requirements including raw materials, parts, and components, assemblies, packaging, and labeling must be carefully performed. Then, a sequence of runs is planned to aim to find what should be the window of work for the parameters being evaluated. Design of Experiments (DOE) is a very useful technique to help to define operational ranges. At this phase acceptance criteria and sample size are established for both the short and long run, while the extreme values of the operational range (worst-case conditions) are determined, to comply with regulatory demands.
An innovative company has challenged Gsap to determine what parameters should be validated and what would be their levels for a successful mold injection of a Uterus Self Retaining Support (USRS) implant. The melted material was extremely expensive so that the runs needed to be precise. Decisions for trials were taken beforehand based on a very high professional knowledge. The validation team decided that Holding Pressure and Cooling time should be challenged at two levels each, namely High and Low. A methodical DOE took place outcoming the window of work for the acceptable extreme values. The recommended levels were successfully determined for the next phase, the Performance Qualification. Three batches, all within the recommended levels, concluded the PQ.
The example above was completed under 100% acceptance of the items produced. The summary report emphasized the need for monitoring both the cooling time and holding pressure for runs at normal operating conditions.
Yes, this is correct. We all know process instruments must be calibrated, but calibration alone will not suffice inspections. It sounds weird, even fastidious, yet it is easy to understand. Suppose that a part that is produced at high volumes has a Critical to Quality (CTQ) dimension. This variable is manually measured along with production, in three different shifts, by one calibrated micrometer.
Regulations require that the measurement method produces valid results, regardless of the instrument used, variable, or attribute data alike. Back to our example, if we were up to measure the same part at those three shifts, we would expect to get three different results. The theoretical difference might not be significant, but can you prove it? Let’s be aware that the tolerances are not of concern here. At times, you will be required to present supporting documentation showing that measurements or testing methods do not vary more than 10% over shifts and operators, regardless of acceptance. In other words, variations in results should be mostly due to the difference among parts and not vary due to humans or any other factor over ranges and time.
Likewise, for a large staff, an agreement between different QC personnel may as well be questionable.
This is the so-called Test Method Validation (TMV) where the Repeatability and Reproducibility of testing and measurement methods (Gage R&R) are used.
TMV is an integral part of any successful validation for manufacturing processes. Again, our professional support in developing correct protocols and executing them properly will help you overcome the TMV challenge.

If your product has software applications associated with it, you are required to confirm that the software specifications conform to the intended use of the product and they fit the user’s needs. Unless there is an in-house specialist on Software Validation this is definitively the place to outsource support.
Electronic Records and Signatures, data integrity and workflow, accessibility, audit trail, and retrieval capabilities are a few of the qualifications needed.
A Bio-Tech company in the cell-growing for blood disorder market had a large number of QC lab devices, each with its own dedicated software application, turned to Gsap for Software Validation support. Their product was stored at deep freeze. The analysis performed by each device, the results, and the generated reports for batch release were backed up in the cloud. Besides the qualifications needed for all temperature conditions, cyber protection for data became a must. But how to validate software that is working only with cloud-generated files? We started with a GAP Analysis for the computerized Systems for each device. Next, the work focused on a Criticality Assessment and Software Installation issues. Finally, we approached Configurations, Security Measures, Remote Storage, and Retrieval features, Data Work Flow, Data Integrity, Audit trails, Readability, and Disaster recovery. As a result, qualifications have fulfilled the requirements not only for the 21 CRF Part 11 but also for the Annex 11 of the Eudralex EC Public Health and Risk Assessment. ALCOA+ requirements were also covered.
We found a way to cope with one of the fear-most requirements: Statistical techniques. It is easier to think of it as self-defense for your project. In this way, the sampling rationale becomes even helpful.
Suppose you have adopted the ANSI Z1.4 for inspections. Now you can state the purpose of the acceptance plan, how much is routinely accepted 95% of the time, how much is expected to be rejected, and so on. Tables and Operational Characteristic curves (see below) are of easy access and ready for your ease. It seems everything is Ok.
But the ANSI is mostly used for lots that have been already produced. It looks to the past. What happens if you are going to validate a batch to be produced in the future? How to decide which is the appropriate success criterion looking to the future? You will need to assume a standard deviation of the upcoming sampling and hope variation does not exceed it. Well, how big must the sample size be for a high degree of confidence? What reliability do you desire for your product? After all, decisions are based on a sample. What happens if there are multiple runs and different batches? Do they share homoscedasticity (common variance)? The answers are not so simple.
Cp and Cpk indexes are common ways to validate manufacturing by simply proving the capability of a process. However, they require samples to be originated from a normally distributed population, an issue frequently overlooked. Can you prove the normality of your data? How many samples do you need for a minimum Cpk of 1.33? Are there outliers? What to do with them? How to interpret the p-value obtained? These are some of the questions concerning the statistical side of Validations.
Our support starts by analyzing results. If they do not come from a normally distributed population, we will search for the appropriate distribution to work with it. Occasionally data transformation may be of help. Those issues are important to decide the overall validity of the analysis. They require some expertise, but whatever the case is, we will help you comply with the statistical requirements, select the correct sample size, make sense of data, and feel comfortable with the numbers.

Benefits and why is Process Validation so important?
First of all, because it can be enforced on you. It’s the law. The Code of Federal Regulations (CFR) are not recommendations but rules that must be followed.
Additionally, there are several advantages for validating manufacturing processes in the medical device industry.
●It ensures your equipment operates correctly and is maintained properly. This means immediate savings, less downtime, and product quality benefits.
●In the long run, it reduces inspection and correction costs. Validated processes shall be monitored and controlled so that your production and products are safe and reliable over time.
●Processes are understood by the staff. It makes good business sense when you and your staff comprehend and control the processes. A knowledgeable staff means your business is competitive and ready to cope with any of the day-by-day challenges.
Validated processes under regulatory compliance shall be one of the ultimate goals for your business. This may be the right moment to improve existing Validation procedures, or if you are new on the subject, to correctly introduce Validation, alongside professional help.
Improving your staff’s capabilities for the development and execution of qualifications is also a way to go with Gsap seminars and courses. For more information, please contact us.
Now is your turn.
This article was prepared by:

Jose (Yossi) Chvaicer, M.Sc.
Senior Validation and Quality Engineer
We are pleased to attend together with SE Pharma for the upcoming IPL Forum 2021- supply chain management conferences for the healthcare and food-tech industries!
The conference was held on 20.10.2021 at the Avenue Congress Center.
Gsap experts provide professional lectures:
The Annual Healthcare Industry Supply Chain Management Conference-IPL Forum 2021
Opening seat
9:20 – Implementing GDP in Israel – Regulatory Challenges and Key Insights from Audits:
Dr. Sigalit Arieli Portnoy, CEO

Gsap provides guidance to the Israeli Medical Cannabis industry: start-Up companies as well as mature companies with established factories.
Check us out on page 3!
For the PDF version press the “HAARETZ” link below:

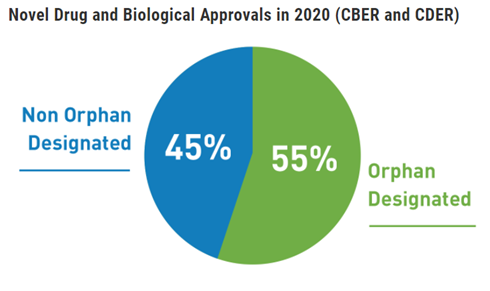
Over 300 million people are living with at least one of identified rare diseases around the world. Rare diseases currently affect 3.5% – 5.9% of the worldwide population. 72% of rare diseases are genetic, 70% of those genetic rare diseases start in childhood.
The fact that there are often no existing effective cures offers development products regulatory priority, this benefit together with appropriate investigational planning can lead to fast drug approval.
This newsletter covers:
●A disease that affects a small percentage of the population. About half of the people affected by rare diseases are children. Most rare diseases are serious or life-threatening. Patients with rare diseases may have no available therapies for the disease.
●An Orphan Drug is a drug for rare diseases. Orphan drugs follow the same regulatory development path as other pharmaceutical drugs. However, the drug can be tested on fewer patients to get approval.
The drugs for rare serious or life-threatening disorders with unmet medical needs may qualify for expedited approvals.
Regulatory priority addressed to unmet medical need
The regulations are intended to speed the availability of new therapies to patients with serious conditions, especially when there are no satisfactory alternative therapies while preserving appropriate standards for safety and effectiveness. The regulatory agency such as FDA recognizes that certain aspects of drug development that are feasible for common diseases may not be feasible for rare diseases and that development challenges are often greater with the increasing rarity of the disease.
●The agencies call for earlier attention to drugs that have promise in treating serious or life-threatening conditions.
●Encourages early consultation for sponsors to plan efficient trial design.
●Applies flexibility in situations to address particular challenges posed by each disease
How to Define the rare disease trial population
●Broad inclusion criteria can allow identification and better characterization of disease phenotypes for which therapy development may be more needed.
●The use of enrichment strategies such as demographic, pathophysiologic, historical, genetic or proteomic, clinical, and psychological characteristics can help to demonstrate treatment effectiveness.
●Choosing patients with a greater likelihood of having a disease-related endpoint event or a substantial worsening in condition.
●Choosing patients more likely to respond to the drug due to their physiology or disease characteristics and /or disease subtypes.
●Include pediatric patients with rare diseases in the study to develop data on the full range of people with the disease.
Sponsors developing drugs for rare diseases face many challenges. These may include the small number of disease-affected individuals, lack of understanding of the natural history of the disorder, lack of precedent for drug development (e.g., established clinical endpoints, validated biomarkers), phenotypic heterogeneity, and the need to conduct trials in pediatric populations.
●Consider the benefits and risks of the drug.
●Consider the seriousness of the disease and if there is an unmet medical need.
●Anticipated safety issues and trial stopping rules, halting rules, and patient early termination.
●Plans for an independent data monitoring committee (DMC).
●Inclusion of patient perspectives in the drug development plan.
●Plans to conduct extension studies to evaluate longer-term safety and durability of effect.
●Considerations related to novel endpoints including the development of clinical outcomes assessments e.g., patient-reported, observer reported, clinician-reported, performance outcome measures.
●Plans for pediatric studies must comply with appropriate regulatory and ethical requirements, including the additional safeguards for children.
Use of biomarkers
●Predictive biomarkers may be used for proof-of-concept.
●Drugs that are intended to be used in a biomarker-defined subtype of patients may require a companion diagnostic that shall co-development with the drug. Companion diagnostics are tests that provide information essential for the safe and effective use of a corresponding drug,
●Pharmacodynamic/response biomarkers allow guide dose-response for more precise dose-finding for therapeutic modalities. Biomarkers can be used to monitor specific parameters and adjust doses so that the biomarkers are kept within specific ranges.
●A response biomarker can be used to show that a biological response has occurred in an individual who has been exposed to a medical product, therefore, provide supportive evidence of efficacy.
A natural history study is a pre-planned observational retrospective or prospective study that follows a group of people over time who have or are at risk of developing, a specific medical condition or disease. A natural history study collects health information in order to understand how the medical condition or disease develops and how to treat it. A natural history study can be submitted as a baseline, in absence of a concurrent comparator, demonstrating the disease course for untreated patients along with data that charts the disease course of patients given the proposed therapy to show how the natural progression is changed or perhaps halted by the therapy.
Retrospective natural history studies most commonly use information in existing medical records can provide quick data.
Prospective natural history studies collect and analyze new data generated from identified patients at specified time points after the natural history study has been initiated provide systematically and comprehensively captured data.
Choose sufficient study duration to capture clinically meaningful outcomes in course of the disease.
Be specific when formulating your research objectives, select potential prognostic characteristics, and disease features that may help formulate a sensitive clinical endpoint
Collect data from clinical examination findings, laboratory measurements, imaging, reports of patient functioning and feeling include the standards of care and concomitant therapies.
Include patients across a wide spectrum of disease severity and phenotypes.
Use standardized collection methods and medical terminology to enhance the value and usefulness of natural history study data.

●For many rare diseases, well-characterized efficacy endpoints appropriate for the disease are not available.
●Endpoints should be selected base on the range and course of clinical manifestations associated with the disease, the clinical characteristics of the specific target population and the aspects of the disease that are meaningful to the patient and that could be assessed to evaluate the drug’s effectiveness.
●Biomarkers can be used as a surrogate endpoint that is considered reasonably likely to predict clinical benefit when analytical and clinical validation of the biomarker test is confirmed by the regulatory agency before starting the clinical trial. Endpoints that are considered reasonably likely to predict clinical benefit, even if not well-established may be used as a basis for accelerated approval for treatment of serious or life-threatening diseases.
●Exploratory evidence from phase I and II trials helps to choose the dose and timing of endpoints evaluation in the advanced stages of the clinical development program.
●When the primary endpoint is clinically meaningful but susceptible to individual interpretation, the trial may benefit from having additional supportive secondary endpoints (e.g., laboratory measurements).
●The validity, sensitivity, reliability, or interpretability of an endpoint may be different for patients with mild or severe forms of the same disease.
Efficacy vs Effectiveness
The term efficacy refers to the findings in an adequate and well-controlled clinical trial or the intent of conducting such a trial and the term effectiveness refers to the regulatory determination that is made on the basis of clinical efficacy and other data.
Studies in healthy subjects may determine which factors influence a drug’s disposition or pharmacodynamic effects, dedicated clinical trials that inform dosing and usage instructions in the target population with a rare disease may be limited. The information from such studies and analyses can inform trial design and serve as supportive evidence of effectiveness. Data generated from such studies and analyses can efficiently optimize conditions for drug use e.g., dose, schedule, patient selection, etc.
In rare disease drug development, given the limited number of available patients, it is crucial to standardize the collection and handling of data to ensure quality and interpretability.
Increased measurement variability reduces statistical power.
Safety
A smaller number of patients may be acceptable when the intended treatment population is small.
Many rare diseases are genetic in origin and characterized by more than one phenotypic subtype. Prevalence estimates should include all phenotypic subtypes of a disorder anticipated to respond to the investigational drug.
Natural history studies can help distinguish drug-related adverse effects from underlying disease manifestations.
Many rare diseases severely affect children, and for diseases that affect both children and adults, sponsors should explore the early inclusion of pediatric patients in clinical studies.
Whenever ethically and practicably feasible, to facilitate interpretation of adverse event causality, especially with respect to the incidence and severity of adverse events that could be a manifestation of the disease under study.
A trial protocol with a safety cohort running parallel to the efficacy trial would include patients with the disease who investigators think might benefit from the investigational drug but who do not meet all the registration trial eligibility criteria. Such patients can be enrolled in the trial, avoiding the need for a separate trial and protocol. However, these patients are not randomized and are excluded from the efficacy analysis.
References
●Rare Diseases: Common Issues in Drug Development Guidance for Industry, January 2019
●Rare Diseases: Early Drug Development and the Role of Pre-IND Meetings Guidance for Industry, October 2018
●Enrichment Strategies to Improve Efficiency of Drug Development, May 2018
●Guidance for Industry Expedited Programs for Serious Conditions – Drugs and Biologics, May 2014
●Patient-Focused Drug Development: Collecting Comprehensive and Representative Input, June 2020
●Patient-Reported Outcome Measures: Use in Medical Product Development to Support Labeling Claims, December 2009
●Providing Clinical Evidence of Effectiveness for Human Drug and Biological Products, May 1998
●Patient-Reported Outcome Measures: Use in Medical Product Development to Support Labeling Claims, December 2019
I●n Vitro Companion Diagnostic Devices Guidance for Industry and Food and Drug Administration Staff, August 2011
●BEST (Biomarkers, Endpoints, and other Tools) Resource, FDA-NIH Biomarker Working Group, December 2016
This Newsletter Prepared by:

Inna Grau, M.Sc. Clinical Trial Manager
The food industry is constantly evolving and we are exposed to the need for new ingredients and products for food and drink. In order to sell food or food products containing components that are not approved for use according to the regulations in Israel, a “Novel Food” approval must be obtained from the National Food Service at the Ministry of Health.
This newsletter will cover the following topics:
Novel Food is defined as food that had not been consumed to a significant degree by humans in Israel before 19 Feb 2006, when the first Regulation on novel food in Israel came into force. ‘Novel Food’ can be newly developed, innovative food, food produced using new technologies and production processes, as well as food which is or has been traditionally eaten outside of Israel.
In order to be categorized as “Novel Food”, the food should belong to one of the following groups:
Examples of Novel Food that was approved to use in Israel include Docosahexaenoic acid (DHA) and Eicosapentaenoic acid (EPA)- rich oil extracted from Schizochytrium sp. microalga, agricultural products from third countries (chia seeds), food derived from new production processes (UV-treated milk) and 2′-O-Fucosyllactose produced by genetically engineered Escherichia coli bacteria.
Cultured meat is an in-vitro culture of animal cells through tissue engineering. These cultured animal cells are grown in a controlled laboratory environment to mimic specific cuts or parts of meat that would be traditionally sold in the market. In the EU, cultured meat would be regulated by the Novel Food Regulation (EU Regulation No 2015/2283) because “food consisting of, isolated from, or produced from a cell culture or tissue culture from animals, plants, micro-organisms, fungi or algae is considered one of the novel food categories listed in the regulation.” Due to growing trade with the European Union (EU), the Israeli food legislation and standardization system are increasingly harmonized to European standards. Similar to Europe, in Israel, cultured meat will fall under the Novel Food category and will be evaluated under the Novel Food premarket authorization process. To date, the application for cultured meat approval by the National Food Service has not been submitted yet in Israel.
The National Food Service at the Ministry of Health is responsible for assuring the safety, quality, and authenticity of food for consumers. This is the regulatory agency responsible for the development of food standards and regulations dealing with foods sold in Israel. The agency is also in charge of imported food licensing.
As detailed in the 004-08 directive for Novel Food implemented since the beginning of 2006 and published on the National Food Service website, any food that can be categorized as Novel (including genetically modified food) undergoes a thorough evaluation process by the National Food Service prior to its market authorization including aspects related to its safety, nutrition, and consumption of the food in Israel. The underlying principles underpinning Novel Food in Israel are that Novel Foods must be:
Food that was authorized as Novel food will be listed in the Novel food/food components approved list published on the National Food Service website.
Novel food or components of Novel food (including genetically modified microorganisms) must undergo rigorous safety assessments by a team of experts. It must meet all requirements and tests and be safe to use and consume (case-by-case) prior to its marketing.
Safety evaluation of genetically engineered food is related mainly to possible risk factors affecting human health consuming the food such as:
An engineered food whose genetic alteration causes an unwanted effect, such as increased toxicity or allergens, is not approved.
To date, the engineered food products that have been independently tested and approved by various food authorities around the world have not been found to have health concerns.
Novel Food Pre-Marketing Authorization Process:
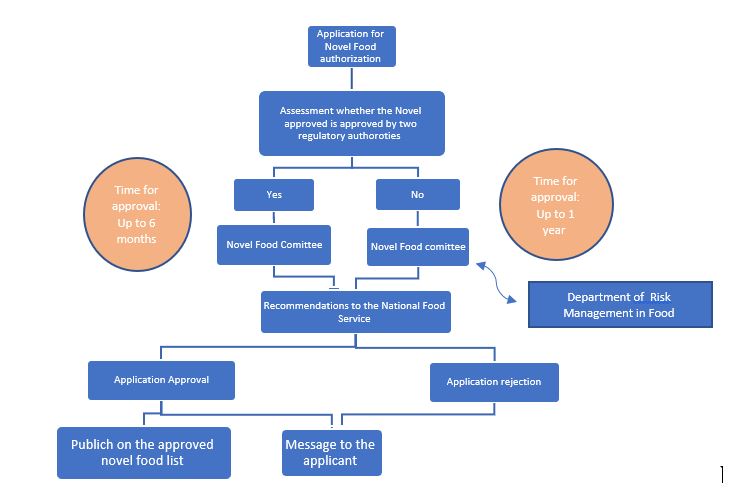
The Public Health (Food) Protection Law – 2015 which came into force in 2016, enshrines the authority of the Ministry of Health to establish safety provisions and regulations regarding the import, sale, and production of Genetically Modified Food (GMO). The main innovation is that the law not only anchors the regulatory authority of novel food safety but also gives the Minister of Health the authority to prescribe provisions regarding its labeling.
In addition to the Public Health Protection Law, there are a number of specific food regulations such as Public Health Regulations (Food) (Gluten Marking), Public Health Regulations (Food) (Marking a Breast Milk Substitute), Public Health Regulations (Food) (Food Additives), Public Health Regulations (Food) (Pesticide Residues), and the Public Health Regulations (Food) (Nutritional Labeling).
On December 25, 2017, the Israeli parliament’s Labor, Welfare and Health Committee approved new regulations for the Protection of Public Health (Food- Nutritional Labeling) which entered into force on January 1, 2020. Currently, Israel has no governmental policy on the labeling of Genetically engineered food products (see below).
Marketing and labeling of genetically modified food products have caused public turmoil around the world.
The legislative situation in the world and in Israel:

A regulation that legally sets the guidelines regarding Novel Food including genetically modified food and its labeling is in the final stages of legislation. Each novel food before its approval undergoes a risk assessment that includes aspects related to the safety, nutrition, and consumption of the food according to a new food registration procedure that has been implemented since the beginning of 2006 and published on the Food Service website. With the entry into force of new food regulations, the obligation to label genetically modified food components will apply, in addition to a safety examination as has been done to date.
The topic of labeling has become an important topic in discussions about engineered foods all over the world. Government ministries, consumer organizations, and food marketing entities in Israel and around the world believe in the consumer’s right to know whether they have used genetic engineering technology in food production or components from it and therefore require the labeling of these products. In Europe, there is extensive activity on the subject and there are clear requirements for labeling genetically modified food, which are regulated by relevant regulations. In contrast, currently in the U.S. and Canada, it is believed that labeling is not necessary because these foods undergo safety assessment prior to marketing, by a team of experts advising government officials responsible for food control, and only products that are safe to use are authorized for marketing.
In Israel, the Food Service adopts the approach that believes the public has “a right to know” and works to regulate the issue of food labeling that has been legally genetically modified. Once installations will pass, it would create a mandatory labeling requirement for food items that contain genetically engineered ingredients.
Health Vs. functional claims labeling on Novel Food products
In the USA a health claim (i.e a statement about a relationship between food and health) labeled on food products (including Novel Food) is not allowed unless it was approved by the FDA. The functional claim of the food is allowed to be added to the label. The functional claim must be correct but is not approved by the FDA (a disclaimer stating that the claim was not reviewed and approved by the FDA must be shown on the label).
In Europe, there is no differentiation between functional and health claims. Each claim labeled on the food must be correct, established with scientific proof demonstrating the direct relationship between the claim and the food function, and approved by EFSA (European Food Safety Authority).
In Israel, no claim for food is currently allowed (except for phytosterols). Once the Novel Food installations will pass, functional claim labeling for food will be allowed- for Novel Food only.
This Newsletter Prepared by:

Tsufit Gross, Ph.D.
Pharma and Biotechnology Regulation Project Manager
What is reality?
In the past, the answer to this question for most people was very obvious- it is the reality that we can approach with our physical senses. Today, the evolution of human consciousness and technology has brought us to a place where we can choose a substitution for the limited physical reality. Today there are alternative and extended realities with Augmented Reality (AR) and Virtual Reality (VR) to name a few.
As with any other innovative technology, the healthcare industry is quick on adopting those extended realities as part of their arsenal and the regulatory bodies have to keep up with the industry. As a matter of fact, the extended realities have so much potential that the regulation harnessing it for its aid also! Just to emphasize the enormous growth of this field, it is estimated that by 2022, the extended realities market is expected to reach $209 billion, which is eight times what it is today [1].
Topics in this article:
Extended Reality (XR) is an emerging umbrella term for all immersive technologies. The ones we already have today—Augmented Reality (AR), Virtual Reality (VR), and Mixed Reality (MR) plus those that are still to be created. All immersive technologies extend the physical reality we experience by either blending the virtual and “real” worlds or by creating a fully immersive experience.
VR and AR devices first made headlines in the consumer sector. Does Pokémon Go rush ring a bell? However, healthcare was quick to spot the potential of the technology, leading to explorations of its use in areas such as doctor-patient communication, surgery training, rehabilitation, and digital cognitive behavioral therapy such as chronic insomnia, for example [2, 3].
AR differs from its most known “relative”, VR since the latter creates a 3D world completely detaching the user from reality. There are two respects in which AR is unique: users do not lose touch with reality and it puts information into eyesight as fast as possible. These distinctive features enable AR to become a driving force in the future of medicine.
There’s plenty of potential for VR and AR in healthcare, but how might people use it, specifically?
Following are just the “tip of an iceberg” for the benefits of extended realities [3, 4]:
Researchers have investigated using VR to educate patients before their surgeries, too. For example, a person can see a digitized version of their brain, along with the problem a surgeon needs to fix and how they will do it.
These technologies could also improve training in medical school. Researchers at Johns Hopkins University embarked on an AR project that could see medical students trade their anatomy apps for AR. An augmented reality tool displayed an internal view of the body on top of a student’s physique. The technology also included a gesture-sensitive user interface, allowing people to interact with the AR representation.
Although the benefits of the XR in the use of healthcare speak for themselves there are also several risks associated with it. If we refer to the regular use of this technology in gaming, for example, Pokémon Go mentioned earlier, by now, it is officially conquered the world. It was reported that on the day when the game was launched, it immediately surpassed the daily time usage of Facebook, Snapchat or Twitter by the average iOS user on mobile phones. The side effects were soon to follow. For example, there were people who quit their job to become full-time Pokémon hunters. In Central Park, herds of Pokémon Go players almost caused a stampede as they tried to capture a rare type of imaginary animal. Car crashes due to the game application became a common phenomenon.
Beyond the daily life risks this technology imposes, there are several specific risks related to the healthcare field. The main risks being:
Surely there are also additional minor risks associated with each specific application.

The first step in determining the extended XR place among medical devices is by determination of its nature by analogy with existing legally regulated categories. Main medical device regulators in the US, Europe, Japan, and other markets have begun addressing some of the challenges through the expansion of guiding documents for the development and evaluation of the Software as Medical Device (SaMD), under which the XR is currently categorized.
The regulatory bodies around the globe are adjusting to the rapidly changing landscape of the technologies available to the end-users. Due to that adjustment, major attention was allocated to the Software as Medical Device field, including revision of the existing regulations and guidance documents and issuing new additional documents to support and expand the regulatory coverage of this field. Furthermore, the US FDA has established the Digital Health Center of Excellence, which goal is to empower stakeholders to advance health care by fostering responsible and high-quality digital health innovation.
The XR technologies are one of the catalysts for the special attention described above. For example, the US FDA held several meetings to discuss virtual and augmented reality in medicine as these technologies are becoming a major part of the healthcare landscape, currently in the surgical training and planning and therapy for pain, anxiety, and post-traumatic stress disorder [8].
Moreover, the US FDA is so interested in making these technologies available to the end-user in the safest and effective way that it supports the companies that develop XR devices by actual involvement in the development process using the Breakthrough Devices Program [9]. For example, in October 2020, the FDA has granted breakthrough designation to AppliedVR’s platform that treats chronic lower back pain and by doing so opened the door for broader use of virtual reality technology in healthcare [10].
The XR technologies outstand also in the FDA’s Software Precertification Pilot Program. One of the nine companies taking part in the FDA’s Pilot Program, Pear Therapeutics, is an XR developing company. The company products are currently FDA cleared with one of the products being the first product submitted through FDA’s traditional 510(k) pathway while also reviewed as part of FDA’s Software Precertification Pilot Program to help build and test FDA’s Digital Health Precertification Working Model 1.0. The company has been working closely with the FDA and volunteered to undergo the first-ever Excellence Appraisal in May 2019, which consisted of an onsite evaluation of the company’s commitment and execution across product quality, patient safety, cybersecurity responsibility, clinical responsibility, and a proactive culture [11]
Pear’s products are also part of a specific group of products designated by the FDA as the Prescription Digital Therapeutic (PDT)- a prescription-only software that delivers evidence-based therapeutic intervention(s) to prevent, manage or treat a medical disorder or disease. Designation of a specific characterization to a group of XR devices represents the emphasis the FDA is putting into these technologies.
The European (EU) regulation is pretty up-to-date as well when it comes to adapting to the rapid advances in technology. The rules on the approval of new medical devices have been updated relatively recently (in May 2017), and the EU is investing a lot in innovation. After a public consultation in 2017, the European Commission confirmed that one of the three priorities of the Digital Transformation of Health and Care in the Digital Single Market is “citizen/patient empowerment with digital tools for user feedback and person-centered care”. This ambition has been reconfirmed with the launch of a new instrument, the Digital Europe program. The European Union will invest €9.2 billion from 2021 to 2027 on several key digital challenges, including (e)healthcare & citizen empowerment [12].
Interestingly, although the XR technologies are covered under the SaMD (Software as Medical Device) umbrella, currently approved devices are verified and validated using the harmonized EN 62304 standard and the FDA Guidance for the Content of Premarket Submissions for Software Contained in Medical Devices. This situation may change though when the new guidance documents and regulation of SaMD will be released and updated.
While no organization is immune to a cyber breach, organizations are expected to secure virtual and physical worlds. This is true especially when the technology is being deployed in critical situations, such as surgical procedures. Rather than viewing these issues as obstacles, meeting them head-on early in the process can help mitigate cyber risks, enable faster deployment and innovation, and reduce brand and reputational risks, both during development by the solution providers as well as during deployment at life sciences and health care organizations [13]
The first step for risk mitigation is the identification of the specific risks. Therefore, risk management should be considered the initially expected standard of care to be evaluated by the regulatory bodies. The risk assessment should begin with the fundamental issues such as identity and authentication in the virtual world that should differ from logging into a laptop with a user name and password to mitigate the risk due to cyber-attack. Embedding risk management throughout the process is crucial for digital transformation.
XR solution developers should incorporate security by design into their product lifecycle, and life sciences and healthcare organizations adopting these technologies should enhance their vigilance by monitoring related technology stacks on a real-time basis through integration to their overall security strategy in areas such as their Security Operations, Vulnerability Management, and Technical Resilience programs, among others. It is essential to integrate robust controls into the product or platform. It is expected by the customers and pursued by the regulatory bodies.
XR submissions are very in-depth and the amount of supporting clinical data. The requirements depend on the specific application of this technology varying from large, multi-centered, controlled studies, through retrospective evaluation, to no supporting clinical data at all.
Soon, we will be able to choose which reality we wish to live in, and regulation is here to make sure we use the benefits of this situation safely and wisely.
This article Prepared by:

Dr. Ella Sheiman, RA & QA Project Manager
Medical device
The Covid-19 Pandemic is one of the greatest challenges modern medicine has ever faced. Hospitals and research labs all over the world are testing many different therapies on coronavirus-positive patients in an effort to find a potential COVID-19 treatment. There are thousands of clinical trials investigating treatments and preventative measures for COVID-19.
This newsletter will review the prominent drugs, the Israeli achievements, and where Gsap helps promote the fight against Coronavirus.
Topics in this newsletter:
Current approaches to COVID-19 therapies generally fall into two categories: antivirals which prevent the virus from multiplying and immune modulators which help the immune system to fight the virus or stop it from overreacting dangerously.
During a public health emergency, such as COVID-19, the FDA can issue an emergency use authorization (EUA) to help make new medications and medical products more available to patients. Having a EUA does not mean that the FDA has approved the medication or product. Rather, the intent of a EUA is to make it easier for patients to receive a new potential treatment when no other options are available.
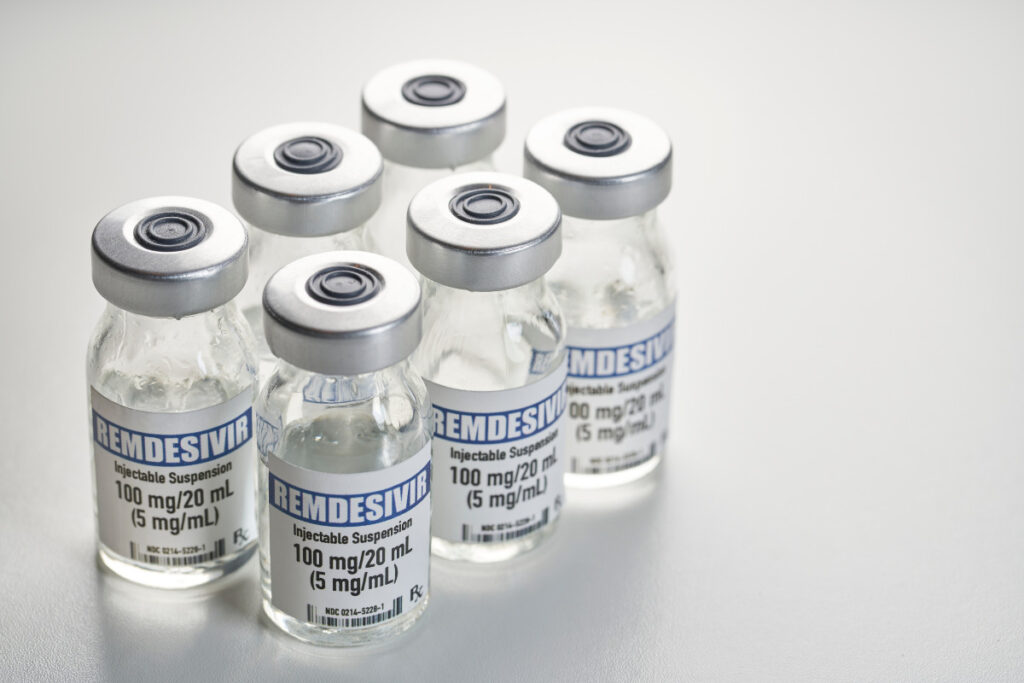
Remdesivir is the first drug to gain approval from the FDA for the treatment of Covid-19 made by Gilead Sciences under the brand Veklury, it works by interfering with the creation of new viruses, inserting itself into new viral genes. Remdesivir was originally tested as an antiviral against Ebola and Hepatitis C, only to deliver lackluster results. But once the Covid-19 pandemic emerged, researchers found that it could stop the coronavirus from multiplying in cells. A large clinical trial was then launched, which found that the drug reduced the recovery time of people hospitalized with Covid-19 from 15 to 11 days. The FDA responded to this data last May by issuing an emergency authorization for Remdesivir’s use in critically ill patients who need supplemental oxygen. In August, they expanded that approval after another study found that patients with less severe forms of Covid-19 seemed to benefit modestly from a five-day treatment course of Remdesivir. The revised approval allows the use of the drug on all patients hospitalized with Covid-19, regardless of how severe their disease is.
Yet many experts remained skeptical of remdesivir’s benefits. They pointed out, for example, that there’s no statistically significant evidence that remdesivir actually prevents deaths from Covid-19. In Nov 2020, the World Health Organization recommended against using remdesivir. Based on a review of all the published trials so far, they concluded that evidence of its benefits is lacking.
Monoclonal antibodies are laboratory-made proteins that mimic the immune system’s ability to fight off harmful pathogens such as viruses. Casirivimab and imdevimab are monoclonal antibodies that are specifically directed against the spike protein of SARS-CoV-2, designed to block the virus’ attachment and entry into human cells.
In November 2020, the U.S. Food and Drug Administration issued an emergency use authorization (EUA) for basiliximab and imdevimab to be administered together for the treatment of mild to moderate COVID-19 in adults and pediatric patients (12 years of age or older weighing at least 40 kilograms) with positive results of direct SARS-CoV-2 viral testing and who are at high risk for progressing to severe COVID-19. This includes those who are 65 years of age or older or who have certain chronic medical conditions.
In a clinical trial of patients with COVID-19, casirivimab and imdevimab administered together were shown to reduce COVID-19-related hospitalization or emergency room visits in patients at high risk for disease progression within 28 days after treatment when compared to placebo. The safety and effectiveness of this investigational therapy for use in the treatment of COVID-19 continue to be evaluated.
Convalescent plasma is frequently administered to patients with Covid-19 and has been reported, largely on the basis of observational data, to improve clinical outcomes. Minimal data are available from adequately powered randomized, controlled trials.
The New England journal of medicine published in February 2021 a Randomized Trial of Convalescent Plasma in Covid-19 Severe Pneumonia. Adult patients with severe Covid-19 pneumonia in a 2:1 ratio receive convalescent plasma or placebo (A total of 228 patients were assigned to receive convalescent plasma and 105 to receive placebo). The primary outcome was the patient’s clinical status 30 days after the intervention, as measured on a six-point ordinal scale ranging from total recovery to death. On day 30 day, no significant difference was noted between the convalescent plasma group and the placebo group in the distribution of clinical outcomes according to the ordinal scale.
Corticosteroids (often called steroids) are used to tamp down inflammation and for conditions such as allergies and asthma. The Covid-19 pandemic brought a new interest in these drugs, and a raft of new clinical trials was launched. In June 2020, the steroid dexamethasone was the first shown to reduce Covid-19 deaths. A study of more than 6,000 people found that dexamethasone reduced deaths by one-third in patients on ventilators, and by one-fifth in patients on oxygen. It may be less likely to help and may even harm patients who are at an earlier stage of Covid-19 infections, however. In its Covid-19 treatment guidelines, the National Institutes of Health recommends only using dexamethasone in patients with Covid-19 who are on a ventilator or are receiving supplemental oxygen.
In September 2020, researchers reviewed the results of trials on dexamethasone, along with two other steroids, hydrocortisone, and methylprednisolone. Overall, they concluded, steroids were linked with a one-third reduction in deaths among Covid-19 patients.
The EXO-CD24 substance, developed at the Ichilov Medical Centre in Tel Aviv, successfully completed its first phase of clinical trials.
CD24 is a small heavily glycosylated GPI-anchored protein. CD24 is a key player in the vast majority of human cancers and also plays an important role in controlling the homeostatic proliferation of T cells. Hence, CD24 can negatively regulate inflammation.
The treatment is a biologic therapeutic agent based on exosomes carrying CD24. The rationale for this treatment is that exosomes overexpressing CD24, isolated and purified from T-REx™-293 cells engineered to express CD24 at high levels, can suppress the cytokine storm and are delivered directly to the target organ (the lungs) using exosomes as a highly body-compatible delivery vehicle. This enables a strong reduction of the required dose and reduces the risk for adverse events.
The treatment was administered to 30 patients with moderate-to-severe symptoms of Covid-19. Twenty-nine of them recovered in up to five days. No placebo was used in the first stage of the trial, and the next phase of the clinical trials will continue to examine the effects and efficacy of the treatment.
During a recent visit to Israel, Greek Prime Minister Kyriakos Mitsotakis offered to have a hospital in Greece take part in clinical trials.

Israeli immunotherapy company Enlivex Therapeutics reported positive interim results of Phase II clinical trial of its Allocetra product in severe and critical Covid-19 patients. The interim clinical results relate to eight Covid-19 patients who were treated with Allocetra, six of whom were in severe condition and two of whom were in critical condition. Seven completely recovered and were discharged from the hospital, after an average of 4.7 days following Allocetra administration. The eighth treated patient in the Phase II study has experienced a clinical improvement following treatment with Allocetra and is currently classified as a moderate/severe condition. The company says that the Allocetra treatment has been well tolerated with no treatment-related serious adverse events. On December 3, 2020, the Company reported positive interim results of Phase II investigator-initiated clinical trial of Allocetra in COVID-19 patients in severe/critical condition.
The interim clinical results relate to eight COVID-19 patients who were treated with AllocetraTM in Phase II clinical trial, six of whom were in severe condition and two of whom were in critical condition. Key results and conclusions from both the ongoing Phase II clinical trial, as well as a previously-reported investigator-initiated Phase Ib study include:
Vaccines typically require years of research and testing before reaching the clinic, but in 2020, scientists embarked on a race to produce safe and effective coronavirus vaccines in record time. Researchers are currently testing 71 vaccines in clinical trials on humans, and 20 have reached the final stages of testing. At least 78 preclinical vaccines are under active investigation in animals.
In November 2020 Pfizer and the German company, BioNTech made history by announcing that their coronavirus vaccine had an efficacy rate of over 90 percent, far surpassing expectations. It was the first time anyone had found such evidence. Just over a month later, in December 2020, the Food and Drug Administration granted it the first emergency use authorization ever given by the United States to a coronavirus vaccine.
In January 2020, BioNTech researchers started molding a genetic molecule called messenger RNA (mRNA) which create the genetic instructions for building a coronavirus protein, known as a spike. When injected into cells, the vaccine causes them to make spike proteins, which then get released into the body and provoke a response from the immune system. In May a clinical trial was started.
In Phase 1 trials, the researchers found that Comirnaty caused volunteers to produce antibodies against SARS-CoV-2, as well as immune cells called T cells that respond to the virus. In July 2020, the companies announced the launch of a Phase 2/3 trial with 30,000 volunteers. In September, Pfizer and BioNTech announced that they would seek to expand the trial to 44,000 participants, and in November 2020, Pfizer and BioNTech released a preliminary analysis of the first 94 cases. Comirnaty has an efficacy rate of 95 percent. While Comirnaty caused no serious side effects, it frequently caused short-lived fatigue, fever, and muscle aches. These impressive results led rapidly to authorizations across the world.
Israel is currently leading the world in vaccination. Israel vaccinated 53.7% of its 9 million inhabitants, with at least the first dose of the vaccine (Until the first of March(. Pfizer and Israeli health officials released new data that shows that the Comirnaty vaccine is greatly reducing transmission, which is one of the most asked questions in the world right now. The Israeli Health Ministry found that the full two doses of Pfizer reduce infection by 89.4% in asymptomatic cases, where there are no visible or tangible symptoms. In cases that bring symptoms, Pfizer seems to work to provide a startling 93.7% level of protection. The vaccine was also 92% effective at protecting people from severe illness after two shots, with a strong 62% protection level after a single dose. Three weeks after the first dose, people reported a 72% level of protection. Scientists expect this percentage to increase over time, as immunity builds in the body.
Johnson & Johnson’s Covid-19 vaccine was endorsed at the end of February 2021 by a panel of experts advising the Food and Drug Administration, clearing the last hurdle before a formal authorization.
Johnson & Johnson’s formulation worked well in clinical trials, particularly against severe disease and hospitalizations, even though it did not match the sky-high efficacy rates of the first two vaccines made by Pfizer-BioNTech and Moderna.
Johnson & Johnson launched a Phase 3 trial in September, which they paused on Oct. 12 to investigate an adverse reaction in a volunteer. The trial resumed eleven days later. Although Johnson & Johnson initially set out to recruit 60,000 volunteers, it capped the trial at 45,000 in December as cases rose.
The Israel Institute for Biological Research announced the start of phase two with a vaccine for the COVID-19 virus (IIBR-100/ BriLife). The study was started with a dose-escalation phase (phase I) during which subjects (18-55 years old) were randomly allocated to receive a single administration of BriLife at low, mid, or high dose or saline or two administrations of IIBR-100 at a low dose, or saline, 28 days apart.
Based on results obtained during phase I, and cumulative phase I data review, the expansion phase (phase II) was started, during which larger cohorts, as well as elderly age subjects, were randomly allocated to receive prime-boost administration of BriLife 28 days apart. The subjects will be followed for a period of up to 12 months post-last vaccine administration to assess the safety and efficacy of the vaccine.
Gsap has the honor to accompany the Israel Institute for Biological Research through the various stages of development and clinical trials.
This Newsletter Prepared by:

Sara Blumenstein Pharma & Biotechnology Regulatory Section Manager








{kind=link}