All companies aim to have high-quality products and services, it is the rare few that actually achieve this desirable goal. The way to achieve this goal is by asking and answering 3 questions:
- Do we have a culture for quality and how is that we know this?
- Do we have a quality product and how is that we know this?
- How can we develop and maintain an efficient and effective QMS?
This article gives you practical tools for answering these 3 questions and demonstrates how Gsap can help you to establish a culture for quality, manufacture quality products, and have a sustainable, efficient, and effective quality system.
Do we have a culture for quality and how is that we know this?
A major survey, conducted by CEB (Corporate Executive Board Company) in 2013 under the topic “Culture of Quality Benchmarking survey”, used the following tool to estimate the culture for quality in In hundreds of organizations.
Gsap can help you implement this simple tool in your organization:
Step 1 – Data collection:
Ask employees in the organization to specify for each of the following 4 statements, whether they agree or disagree with the following statements.
● “I hear quality in the organization”
● ”I see quality in the organization”
●”I feel quality in the organization”
● ”I transfer quality in the organization”
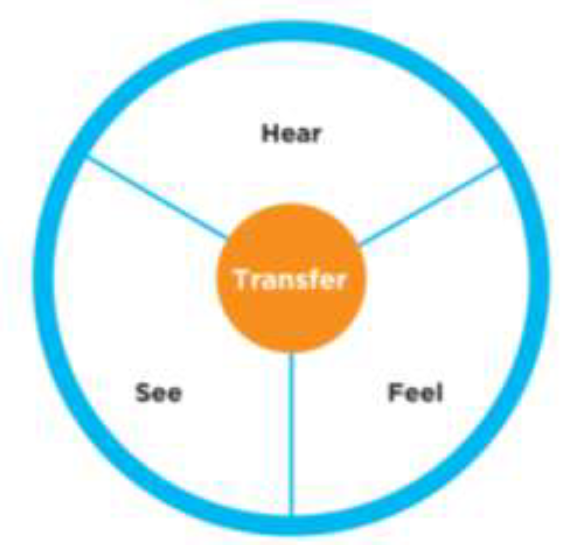
Chart #1 – The cycle of “culture for quality”
Present the data in the following table and calculate the scores:
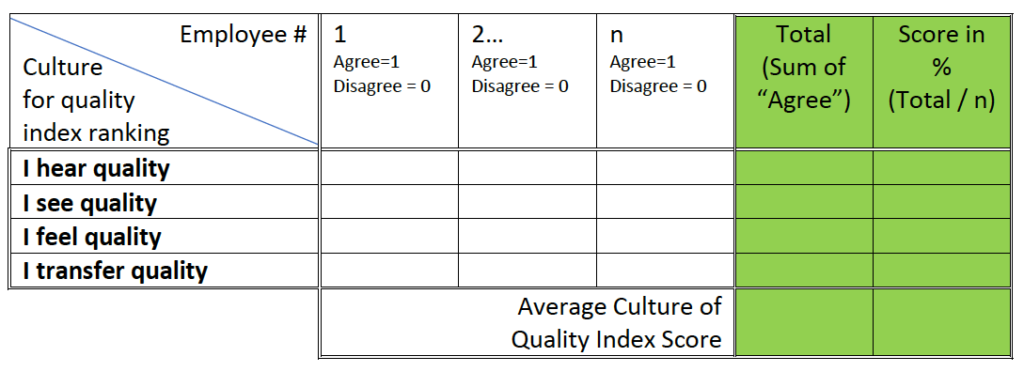
Table #1 – “Culture for quality” survey data collection
Step 2 – Benchmark and Criteria:
Compare your scores with the score received in the CEB survey for companies with high quality/culture for quality.
In those companies the following results were received:
| I hear quality | 90-100% |
| I see quality | 90-100% |
| I feel quality | 45-80% |
| I transfer quality | 90-100% |
Table #3 – “Culture for quality” survey benchmark per quality index
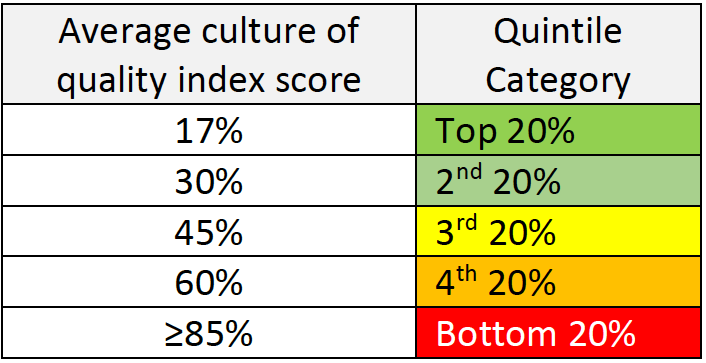
Step 3 – Data analysis and actions to be taken:
If you find that your company is in the 3rd quintile or lower, it is time to define some steps to improve the situation. We, at Gsap, can help you do that by identifying the root causes and building together with you a strategy for change. Some of the root causes for low culture for quality may be:
■ Failure to implement company values from top management to each and every employee
■ Failure to maintain consistent leadership emphasis and help leaders recognize where their actions contradict stated quality policies and objectives.
■ Failure to ensure that quality messages are credible and relevant. For diverse organizations that operate in multiple geographies, this means finding a way to tailor messages without ceding control and being globally consistent yet locally relevant.
■ Failure to demonstrate peer prioritization of quality by generating a social climate that encourages employees to support each other in generating quality ideas, without this effort appearing to be pushed from the top down.
■ Failure to build employee ownership through targeted guidance, setting forth enough guidance to give employees the confidence to act, yet not so much that independent decision making is stifled.
Do we have a quality product and how do we know that?
In 2011 the FDA published their initiative for “Case for Quality”, saying that “Compliance to regulations is not enough…
The goal for the case of quality is to afford patient access to high-quality medical devices”
The aim of this initiative was to arm the industry with practical metrics to implement that meet both the business needs and the product needs so that the Right-First-Time mentality from the initial day of development will return as much as possible.
The FDA defined matrixes in 3 major areas.
Our quality experts in Gsap can help you establish these matrixes, properly collect the data, analyze the results, and set actions for improvement.
Following are some effective Implementation principles
● Manage the Metrics –
Define & control: Collection of raw data, Calculations, Harmonization of definitions, Analysis method, Root cause investigation, etc. to avoid erroneous results that drive wrong decisions.
● Interpret in context –
to understand causes, cross analyze with other metrics and identify how to improve control. Use risk-based approach.
● Apply internally –
Apply within the organization – not compare scores with other organizations out of context.
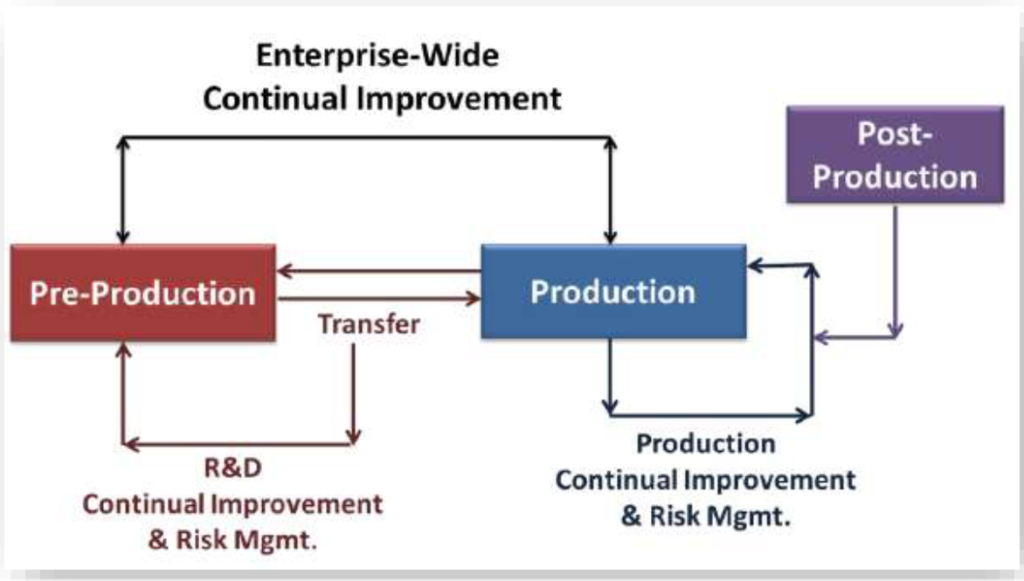
Pre-Production Metric:
Total # of changes (product & process across projects)
________________________________________________________
Total # of projects
Goal:
To drive the Right-First-Time (RFT) mindset in the R&D phase such that post-design transfer changes (after design freeze) due to inadequate product /process development are not needed.
Calculation:
►Only include changes required due to the inadequate product or process development
► Harmonize the definition of “changes” across organizations and classify the significance of varying levels of changes based on risk.
► Determine what constitutes a project (“project” encompasses all aspects of the product being transferred to production).
► Ideal to measure the changes required during (1) Design transfer (after design freeze), (2) Production, (3) On-market
Production Metric
Total # of units manufactured right first time (within or across lots)
__________________________________________________________
Total # of units started
Goal:
To gather nonconformance information during production operations related to inadequate product/process development.
Analysis of this information will enable us to assess the effectiveness of the development process and improve it.
Calculation:
►Include all non-conformances initially (to get a first past yield) & Scrapes
►Triage the root causes
►Track and trend on a rolling basis
►Ensure terms are defined consistently across products and sites
►Raw materials/component failures are not to be included.
Post-Production Metric
Here we have several matrices:
(3) Unplanned Service Records (% out of total units in service)
(4) Installation failures (% out of total installations)
(5) Complaints (% out of total units sold, including non-safety complaints)
(6) MDRs (% of units with “Medical device reporting”, out of total units sold)
(7) Recalls (% of products that had to be recalled, out of total units sold)
Calculation & Analysis
►Step 1: Calculate and track
each of the post-production metrics. Identify trends, and have predetermined action limits.
►Step 2: Use advanced calculations to incorporate product risk profile information into the calculation of each indicator (see table below).
►Step 3: Use comparative analyses to conduct through mechanisms such as dashboard.
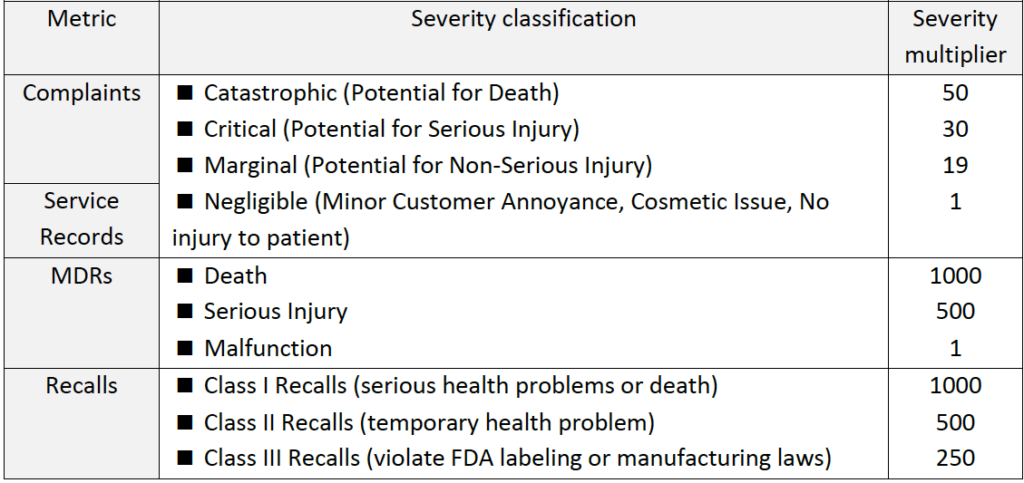
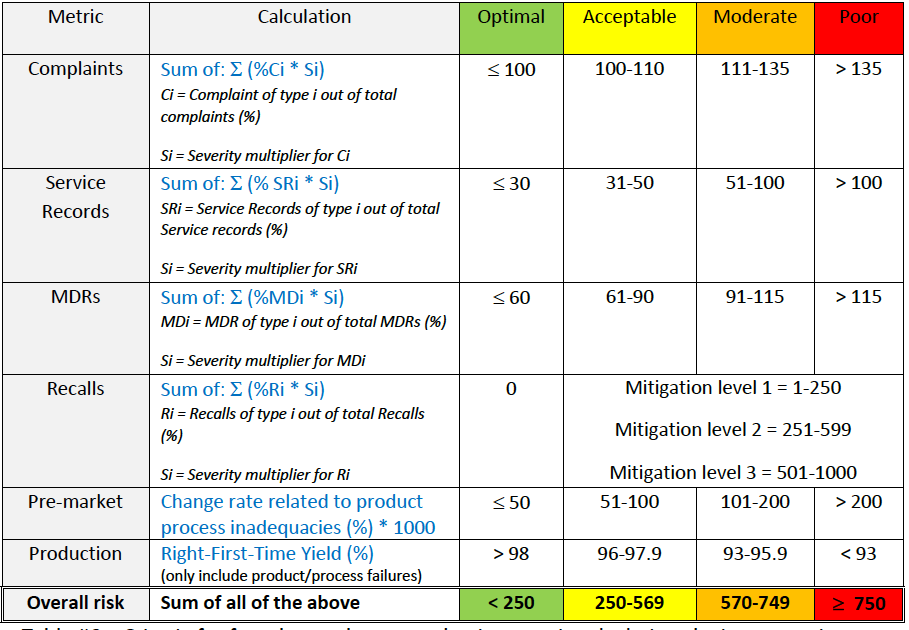
Table #6 – Criteria for advanced post-production metric calculations by incorporation
of product risk
Finally:
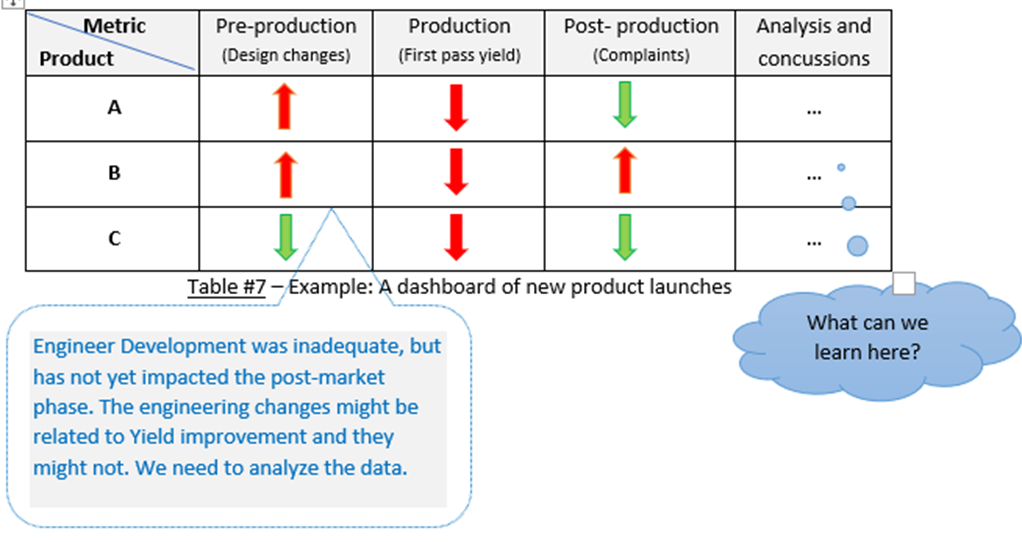
You can incorporate pre-production & production metrics with post-production.
This can give you an enterprise-wide view of the risk to product quality
How can we develop and maintain an efficient and effective QMS?
An effective QMS is a QMS that achieves its goals and objectives. But, in order to have a sustainable quality culture, being effective is not enough – we also need to be efficient.
Having an efficient QMS means a system that meets the company’s needs. A smart and “thin” and agile system where you have zero unnecessary bureaucracy.
Gsap quality experts can help you to build the QMS that is right for you!
Our quality services include the following:
| Culture for quality | Building a culture for quality, based on company values and substantial-quality metrics | |
| QMS procedures | Writing procedures that are suitable for your company | |
| Document and Record controls | Building DHF, DMR, DHR, etc. Electronic QMS (eQMS) by our sub-company ALMOND | |
| e-QMS | Implementation of a full QMS on an electronic platform (ALMOND) | |
| QMS improvements | Management of CAPA, NC, complaints, etc. | |
| Quality Training | Training for all elements of the QMS, including training for employees, quality engineers, Q&R managers, and management. Public and tailored training by our sub-company Gstudy | |
| Quality Audits | Internal audits, Mock -Audits, supplier audits, and audits Audits by ISO 13485:2016, EU-MDR 2017/745 and MDSAP | |
| Preparations for 3rd party audits | Gsap Q&R experts are certified as lead auditors and can help you to be prepared for 3rd party quality audits from regulatory authorities to due-diligence | |
| Total quality management services | Gsap Q&R expert can help you to manage your quality and regulatory activities, including providing guidance to your PRRC (person responsible for regulatory compliance) | |
| Design controls | Building the right flow and steps for full design controls, from concept to getting your regulatory approvals and marketing | |
| Risk management | Implementing risk management procedures according to applicable regulations and standards (such as ISO 14971:2019 and ISO 24971:2020) including the incorporation of usability risks and other related risks, using applicable tools such as FMEA and FTA | |
| Supplier controls | Evaluation and selection of suppliers and ongoing supplier control. | |
| Production controls | Environmental controls, equipment qualification (IQ, OQ, PQ), process validation, measuring devices controls including TMV (test method validation) | |
| Statistical techniques | Including statistical acceptance sampling (based on international standards), statistical process control (SPC), evaluation of process capabilities (such as Cpk and Ppk) determination of sample size for product verification, product validation, and process validation |
Gsap experts are here for you, to help you establish a culture for quality, build quality products, and have a sustainable, efficient, and effective quality system!
This Article Prepared by:

Marina Lebel, B.Sc, CQE
VP Medical Device