The Digital Health Revolution is happening all around us and it is both exciting and terrifying. As a member of the Medical Device community, we have a responsibility to our patients, our families, and our shareholders to follow the current best practices. To date, the standards and regulations have not been able to keep pace with the needs of the patients and the industry that satisfies these patient and user needs.
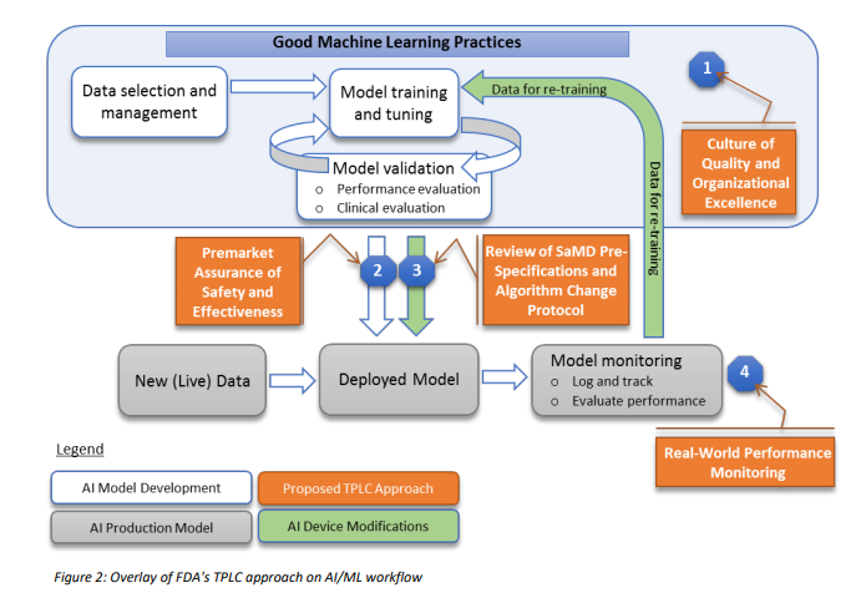
Many regulatory agencies including the FDA are talking about forming committees that will codify the “current best practice”, often referred to as GxP or “Good [insert name] Practice”. In January 2020, the FDA released a discussion paper titled US FDA Artificial Intelligence and Machine Learning Discussion Paper,in this paper, the agency proposes a framework for Good Machine Learning Practices, and below is a visual representation of the GMLP workflow.
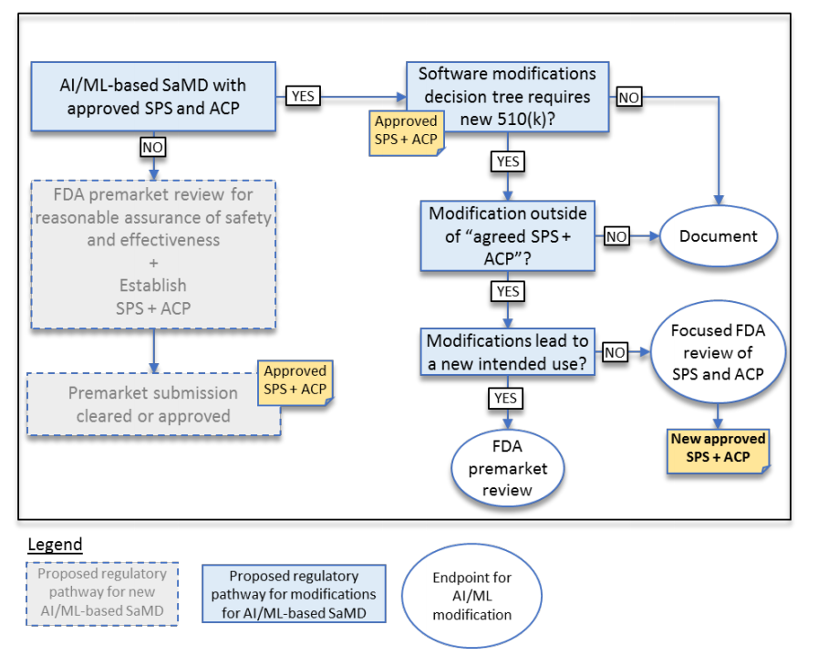
In addition, to address the critical question of when a continuously learning AI/ML SaMD may require a premarket submission for an algorithm change, this discussion paper proposes a framework for modifications to AI/ML-based SaMD.
To date, the FDA has cleared or approved several AI/ML-based SaMD however these have only included algorithms that are “locked”. The power of many of these AI/ML-based SaMD lies within the ability to continuously learn, where the adaptation or change to the algorithm is realized after the SaMD is distributed for use and has “learned” from real-world experience. Following the distribution, these types of continuously learning and adaptive AI/ML algorithms may provide a different output in comparison to the output initially cleared for a given set of inputs.
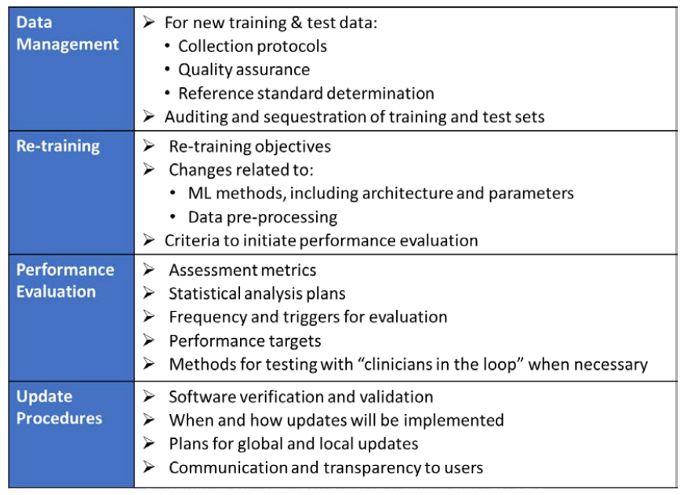
To adapt to this, the FDA is proposing a principle of a “predetermined change control plan.” The predetermined change control plan would include the types of anticipated modifications based on the retraining and model updating strategy, and the associated methodology – referred to as the Algorithm Change Protocol (ACP) – to be used to implement those changes in a controlled manner that manages risks to patients, see below for an outline of the main components of an ACP:
Back in July 2020, the FDA released a Final Guidance Document looking at what happens when the same medical product has both functions covered by the FDA and functions not covered by the FDA. The uniquely unclear guidance titled Multiple Function Device Products: Policy and Considerations look at what happens when a single product with multiple functions have some that are “regulated” and require FDA review, clearance, or approval other functions that do not require FDA involvement (other regulations like FCC or CE standards for electronics, RoHS, WEEE, UL and other TLA’s may be required).
A “function” is a distinct purpose of the product, which could be the intended use or a subset of the intended use of the product. A product with an intended use is to store, transfer, and analyze data has three functions: (1) storage, (2) transfer, and (3) analysis.
While storage & transfer may not be considered requiring regulatory oversight, the addition of analysis and the type of analysis may require FDA involvement. To make things even more complicated, the FDA has issued guidance that indicates while not “fully ok” the FDA does not intend to focus its regulatory oversight on some devices that pose a low risk to patients for more on this ever-growing category see the FDA’s guidance “Policy for Device Software Functions and Mobile Medical Applications” and “General Wellness: Policy for Low-Risk Devices.”
Many CEO’s will attempt to push themselves into this “low-risk category”, as with most things, the agency has discretion, but with software being the leading cause of recalls in the US, the FDA will be waiting to investigate any complaints and will be looking to punish those that have not followed the guidelines to the agencies liking.
So how do we determine if the “non-FDA-regulated” or as the FDA likes to call it the “other function” impacts on the “regulated” feature? Start with 2 questions, and answer them as if you were working for the FDA:
1) Is there an impact on the safety or effectiveness of the “regulated” feature as a result of the “other function?”
if yes,
2) Could the impact result in increased risk or have an adverse effect on the performance of the device function-under-review.
I would very much like to say that from the FDA’s perspective the following is true:
if the “other function” shares code then the answer to both is yes.
if the “other function” shares the same output screen or graphical user interface, the answer to both is yes.
However, there are always exceptions and those edge cases are why we sometimes need to speak with a member of the Gsap digital health team Regulatory Review Team to confirm.
Below are a number of relevant examples from the guidance for your consideration, while some may enlighten, others may confuse, but that is the art of regulatory science.
This Newsletter Prepared by:
Yaron Eshel,Q&R project manager
Medical device, Digital Health Discussion Team
For more information about our Digital Health services visit:
The Nobel Prize for Chemistry was awarded this year for the invention of Genetic Scissors: a tool for rewriting the code of life.
Emmanuelle Charpentier and Jennifer A. Doudna have discovered one of gene technology’s sharpest tools: the CRISPR/Cas9 genetic scissors. Using these, the DNA of animals, plants and microorganisms can be changed/edited with extremely high precision.
This technology has had a revolutionary impact on the life sciences, is contributing to new cancer therapies and may make the dream of curing inherited diseases come true.
Since the discovery of CRISPR/Cas9, the research of this tool has led to a blooming landscape of pre-clinical and clinical studies in humans. FDA considers any use of CRISPR/Cas9 gene editing in humans to be gene therapy, thus requiring extensive regulatory efforts in order to bring such products from early concept to clinical application.
Gene therapy products are regulated by the FDA’s Center for Biologics Evaluation and Research (CBER). Clinical studies of gene therapy in humans require the submission of an investigational new drug application (IND) prior to their initiation in the United States, and marketing of a gene therapy product requires submission and approval of a biologics license application (BLA).
At Gsap, our team of Advanced Therapies experts is excited to be at the frontier of this field, with a unique portfolio of process development, preclinical, clinical and regulatory services, assisting our clients to bring gene-therapy products from early POC to realization into clinical use.
If you develop a gene-editing product, do not hesitate to contact us!
Dr. Sigalit Arieli Portnoy, CEO and Founder of Gsap, talks in an exclusive interview about accelerating the development and approval processes of drugs and medical equipment for COVID, including from foreign companies, and reports On the enormous scope of action that accompanies a sense of mission.
Technical Documentation which must include: ●Device description and specification ●Information to be supplied by the manufacturer ●Design and manufacturing information ●General Safety and Performance Requirements (GSPR) ●Benefit-Risk Analysis and Risk Management ●Product Verification and Validation
Classification
Clinical Data
Biocompatibility – the updated ISO 10993-1 is applicable now both as per the MDD and the MDR.
Where to Start?
Classification – classify your device as per the rules laid out in Annex VIII of the regulation.
General Safety and Performance Requirements (GSPR) replaces the Essential Requirements. Fill one out for each device.
Post Marketing Surveillance (PMS) – look at the clinical PMS data you have and make sure you have a plan and a report.
Make sure your PMS is linked to your Risk Management. Clinical data for your own device will be critical for the Clinical Evaluation Report (CER) to establish equivalence and minimize any further Clinical Study requirements.
Risk Management – No longer one document. This should be a group of documents that address all the requirements of both ISO 14761 and the MDR.
Biocompatibility – you must have a plan (Biological Evaluation Plan) and a report (Biological Evaluation Report) that complies with all the updated and latest Biocompatibility ISO standards. (The 10993 series.)
What to do?
1. Classification – This will lead to updating the Declaration of Conformity.
2. GSPR – any gaps identified (validations or issues that have not been reviewed in the past) must be closed. *** See table below for an example of the GSPR vs. the Essential requirements
3. PMS (Post Marketing Surveillance), PMCF (Post Marketing Clinical Follow Up) – start the implementation of the plan as soon as possible and write a summary report of all data collected to date.
4. Risk Management – verify that hazards have been identified and that the clinical risks have been mitigated.
5. Any Biocompatibility testing that has not been done and cannot be rationalized should be performed.
6. Make sure your QMS adheres to the requirements of the MDR – the following requirements are unique to the MDR:
A documented strategy for regulatory compliance
Manufacturers need to verify UDI (Unique device identifier) assignments for their device both before they hit the market and periodically after they are released
A designated person or group of people responsible for regulatory compliance (PRRC)
Documented procedures for clinical investigation and evaluation
Specific PMS documents – PMS plan and a Periodic Safety Update Report (PSUR), including incidents and field safety corrective actions (FSCA)
Management of the supply chain and economic operators
Implantable devices
European data base on medical devices (EUDAMED)
Common specifications
Vigilance
Example of the GSPR vs. the Essential requirements
Wording GSPR (MDR)
Wording Essential Requirements (MDD)
Explanation of the Gap
That the residual risk associated with each hazard as well as the overall residual risk is judged acceptable. In selecting the most appropriate solutions, manufacturers shall, in the following order of priority: 4(a)Eliminate or reduce risks as far as possible through safe design and manufacture 4(b) Where appropriate, take adequate protection measures, including alarms if necessary, in relation to risks that cannot be eliminated 4(c) Provide information for safety (warnings/precautions/ contraindications) and, where appropriate, training to users.”
Information needed to use the device safely.” Within the framework of labeling
Management would need to be “information for safe use” with the quote and where this information is supplied and a usability study to support the information for safe use is valid.
“11. Infection and microbial contamination 11.2 Where necessary devices shall be designed to facilitate their safe cleaning, disinfection, and/or re-sterilization”
13. Information Supplied by the Manufacturer h) if the device is reusable, information on the appropriate processes to allow reuse, including cleaning, disinfection, packaging and, where appropriate, the method of sterilization of the device to be reserialized, and any restriction on the number of reuses.”
The MDR has defined a particular consideration for reusable devices, meaning a clearly stated requirement that must be validated. The MDD does not clearly state a validation requirement for reusable devices, only the requirement to supply the information to ensure safety.
“14.1 If the device is intended for use in combination with other devices or equipment the whole combination, including the connection system shall be safe and shall not impair the specified performance of the devices. Any restrictions on use applying to such combinations shall be indicated on the label and/or in the instructions for use. Connections which the user has to handle, such as fluid, gas transfer, electrical or mechanical coupling, shall be designed and constructed in such a way as to minimize all possible risks, such as misconnection.”
“9.1 If the device is intended for use in combination with other devices or equipment, the whole combination, including the connection system must be safe and must not impair the specified performance of the devices. Any restrictions on use must be indicated on the label or in the instruction for use.”
The MDR has added an additional requirement regarding connections that are user dependent for combination devices.
“15. Devices with a diagnostic or measuring function”
10. Devices with a measuring function”
The inclusion of diagnostic devices within the requirements of a measuring function is new to the MDR, emphasizing the need for validation of measuring functions in diagnostic devices.
“17.2 For devices that incorporate software or for software that are devices in themselves, the software shall be developed and manufactured in accordance with the state of the art taking into account the principles of development life cycle, risk management, including information security, verification and validation.”
“12.1a For devices which incorporate software or which are medical Software in themselves, the software must be validated according to state of the art taking into account the principles of development lifecycle, risk management, validation and verification.”
The MDR has set design and development requirements for software. This means that the D&D software process must be managed by means of documented inputs, outputs, risk assessments, verifications and validations
Gsap will be happy to support you in getting ready for the MDR storm!
Learn how to implement EU GMP standards in medical cannabis. The EU GMP presents the requirements for facility, equipment, documentation, and process control for the production of herbal products.
The FDA takes steps to promote faster access to new medical technologies that are safe and effective: Streamlining premarket procedures, modernizing the 510(k) program, expediting programs for devices that enhance safety and/or resolve critical unanswered medical needs. The recently issued guides include new pathways, expansion of existing programs, and supporting tools such as checklists and assessment worksheets.
Topics in this Newsletter
510(k) Programs
1. Special 510(k) (UPDATED)
2. Abbreviated 510(k) (EDITED CONTENT)
3. Safety & Performance Based Pathway (NEW)
Additional Premarket Programs
4. Humanitarian Device Exempt (HDE) Program (UPDATED)
5. Safer Technology Program (STeP) (NEW)
More FDA Actions
6. Electronic submissions;
7. Benefit-risk determinations;
8. Acceptance review policy (RTA);
9. Accreditation of testing labs (ASCA)
Adapting the repeatedly criticized 510(k) program to advances in safety and technology; providing clearer requirements and more timely review & response, and lowering workload for FDA and costs for the industry.
Let’s take a short tour of some of the ideas and changes that these documents bring about.
What’s new with the 510(k) program?
The 510(k) program accounts for the vast majority of devices that enter the market each year. It is based on the concept of “substantial equivalence”. The FDA now issues final guidance documents for the Special 510(k) Program, the Abbreviated 510(k) Program, and the Safety and Performance Based Pathway, as well as a Format for Traditional and Abbreviated 510(k)s.
1. The special 510(k) program
This program is intended specifically for changes of own existing device, and only if the methods to evaluate changes are considered well established. FDA reviews special 510(k) applications within 30 days of submission. This final guidance expands the scope of the program.
The significant changes in the program:
• Now, under certain circumstances, changes to the intended use may be made.
• Now, under certain circumstances, changes that alter the fundamental scientific technology may be made.
• If performance data is necessary, it is required that methods are well established and that all data supporting substantial equivalence can be reviewed in a summary or risk analysis format.
NOTE: This pathway therefore may NOT be appropriate,
for example, when a novel sterilization method is used; or methods rely on animal/clinical data; or the changes are complex – with many scientific disciplines involved, change from single-use to re-usable, etc. – so that determining the substantial equivalence depends on the FDA’s interpretation of the underlying data.
2. The abbreviated 510(k) program
An alternative approach to the traditional 510(k): Instead of performing a “head-to-head” comparison with a predicate device in order to demonstrate the substantial equivalence, you declare conformity to voluntary consensus standards / special controls / FDA guidance.
This program is NOT new, but merely received a guidance document separate from the special 510(k) program. It includes a short outline of the content required.
NOTE: This pathway is not necessarily “abbreviated” or shorter.
Also, the review time is 90 days, as for the traditional 510(k). However, under some circumstances, when it is difficult to collect data regarding a predicate, it might be more efficient to submit summary reports describing how to reference standards were used or how the device complies with the special controls. Addressing the FDA early to find out which pathway is appropriate, would be a good idea.
3. Safety and Performance Based Pathway
An optional submission pathway, an extension of the abbreviated program. It is intended for well-understood devices, listed by device type, for which the manufacturer would have to meet objective criteria set by the FDA: criteria that would be consistent with safety and performance characteristics of modern predicates. Along with the guide describing the general pathway, five additional draft guidance documents were issued, describing the required performance criteria for specific devices:
Foley catheters
Recording cutaneous electrodes 03Ø06
Orthopedic spinal plating systems
Non-spinal screws & washers
Magnetic resonance (MR) coils
The basics you should know about this pathway:
• It’s not actually different from the abbreviated 510(k).
• The requirements may be more specific, thus clearer to the
manufacturer (that’s a bonus).
• May be suitable when attaining information regarding an
the actual predicate is impossible/impractical.
Additional programs and pathways at focus :
4. The Humanitarian Device Exemption (HDE) Program
HDE program receives updated final guidance, accompanied by the newly revised guidance for Humanitarian Use Device (HUD) Designations. Both follow changes resulting from the “Cures Act”.
The purpose of this program is to encourage the development of devices for the diagnosis and treatment of rare medical conditions.
The main updates:
• Expansion of the HUD designation: The designation is given when the relevant population is up to 8000 individuals per year in the USA, instead of the previous threshold of less than 4000.
• A device may still receive HUD designation in the pediatric population even when the total condition population exceeds 8000 patients if the pediatric population affected does not exceed 8000 patients.
• For diagnostic devices, the threshold applies to not more than 8000 who would be subjected to the diagnosis with the device, including positive and negative results.
• As was before, in order to make use of an approved HUD in patients, oversight of an Institutional Review Board (IRB) in the medical facility is required, and review and approval for the specific individual use are also required. This was previously also required to be performed by the IRB. Now more flexibility is introduced, as an appropriate local committee (ALC), that may be a standing committee with the appropriate expertise, is allowed to review and approve the specific HDE use, and not only the IRB, which may be considered less accessible. IRB facility overview is still required.
• A filing checklist, and tools for the probable benefit-risk assessment are included in the appendices.
5. Safer Technology Program for Medical Devices (STeP)
More action in promoting patient safety:
The draft guidance for this new voluntary program is issued
– complementary to the “Breakthrough Device Program” (BDP), for medical devices that are expected to significantly improve the safety of currently available treatments.
Devices and device-led combination products, that are subject to review under 510(k), De Novo, or Premarket Approval (PMA) submissions, and that are not eligible for the BDP, may enjoy the expedited STeP approach:
• First apply through a Q-submission (pre-sub) to be included in the program;
• Once accepted, go through a prioritized and expedited review of regulatory submissions, with senior FDA management engagement, and assistance with the plan of device and data development.
To apply, the sponsor should explain the innovative approach and how the benefit-risk profile would be improved:
• Does the device reduce the occurrence of a known serious adverse event/device failure mode / user-related hazard or error?
• Does the device improve the safety of another device or intervention?
NOTE: The Breakthrough program which is intended for life-threatening
/ irreversibly debilitating conditions, is mandated by law and would still be prioritized over STeP. This is expressed in the draft with the repeating phrase: “as resources permit”.
Thus, only time will tell if STeP achieves its purpose to open an actual additional expedited pathway.
More FDA actions to streamline, simplify, expedite, clarify…
6. The FDA goes paperless – electronic submissions are on the way
Following the requirements of the FD&C Act, draft guidance regarding electronic submissions was issued. But don’t get rid of your printers just yet – it will take a little while.
The guidance tells us that:
• Application types that would be required to be submitted solely in an electronic format, include 510(k), PMA, De
Novo, Investigational Device Exemption (IDE), HDE, and more.
• Individual, per-type guidance documents will be developed, specifying required formats and implementation timetable.
• IDE compassionate use requests and adverse event reports will be exempt from this requirement.
ATTENTION: In contrast to most guidance documents, which contain nonbinding provisions, this guidance and the following
individual ones – once finalized, will contain both nonbinding AND binding provisions under the statutory authorization of Congress: I.e., sponsors will be required by law to comply with the specified formats & schedules.
7. Clarifying benefit-risk considerations for higher-risk submissions
Two final guides were issued (1, 2), both actually discuss the same subject: determining uncertainty and benefit-risk in PMA, HDE, De Novo, and BDP submissions.
Some key concepts:
• Greater uncertainty may be accepted when there’s a true clinical need: serious/unanswered/small patient population / high anticipated benefit for quick patient access.
• Higher uncertainty greater need for premarket data; Lower uncertainty data shift to post-market may be allowed.
• Post-market data is always required. It is critical to show that it can be collected in a timely manner, especially as risk/ uncertainty increases.
• A new worksheet is provided as an appendix; Recommended as a basis for the benefit-risk assessment process.
• FDA wants patient perspectives – are the patients really willing to accept the risk and the uncertainty?
8. Saving time for FDA reviewers and for sponsors
– acceptance review policy
Final updated “Refuse to Accept” (RTA) guidance was issued for 510(k) and for De Novo submissions. These outline the preliminary procedure the FDA performs to assure administrative completeness of submissions (including timetable), which takes place before the substantive review that assesses the quality of the information.
Some highlights:
• A checklist of required criteria is provided for each submission type (in the appendices).
• FDA will, within 15 days of submission, electronically notify the submitter if: (a) submission was accepted and under substantive review;
(b) the submission was refused PLUS indication of the missing checklist items, or;
(c) FDA did not complete acceptance review on time, thus transferred
submission to substantive review.
• This notification identifies the lead FDA reviewer assigned for the submission.
• The De Novo guidance includes a recommended content checklist as well, relevant also for the substantive review.
• RECOMMENDED: Follow the checklist, complete it, and include it as part of the application. Do NOT leave sections out (if not applicable – a state so & justify). Letters from subject matter experts are helpful.
9. Accelerating review processes by accrediting testing labs to FDA-recognized standards
Draft guidance for The Accreditation Scheme for Conformity Assessment (ASCA) Pilot Program was issued.
OBJECTIVE: Streamline reviews – once a manufacturer presents results from an accredited body, FDA can accept them without a thorough review. The pilot begins with standards for biological evaluation and for electric safety.
Gsap experts will be happy to assist you in choosing the regulatory track!
suits your product and company, and to support you in the submission process.
Gsap seeks solutions to the challenges of the medical cannabis industry to place Israeli companies at the forefront of the world. A snapshot of the company’s growth and the great achievements of its customers
Risk Management is the systematic application of policies, procedures, and practice in various aspects and processes of a Medical Device in order to achieve a safe and effective product.
Risk Management allows the manufacturer to understand the controls and design features needed in their Medical Device. This process ends only at the end of a Medical Device’s life cycle, so even after the Medical Device has been placed on the market, continuous monitoring and identification of new hazards are required.
For Medical Device companies, this process is often complex. This rises not only due to the device complexity of design, materials, production processes, software used, and device function – but also due to the various stakeholders (Clinical aspects, marketing considerations, manufacturing and available technology, suppliers and sub-contractors), each contributes to the way risk is perceived AND CONSIDERATIONS.
ISO 14971 principles are implemented globally in the Medical Device Industry and conforming to the standard requirements is used to show compliance to regulation all over the world.
Towards the transition to MDR (EU Medical Device Regulation) and IVDR (EU In-Vitro Diagnostic Regulation) and the recognition of ISO 14971:2019 by FDA, and other regulatory bodies, Medical Device companies must assess risk management processes and existing documentation.
Gsap is a leading consultancy firm with accumulated decades of experience in the industry of medical devices and pharmaceutical companies. We work with our customers, corporates to start-up companies in R&D, production, and post-market stages. We deliver the shortest pathway, and consider our clients as partners to success, with tailored service and support.
ISO 14971:2019 meets Regulatory Requirements
Risk management according to ISO 14971:2019 (NEW VERSION) is required according to the new MDR (EU 2017/745) which enters into enforcement in May 2021 and IVDR (EU 2017/746) which enters into enforcement in May 2022.
The transition period from ISO 14971:2012 to ISO 14971:2019 for FDA is December 25, 2022. After this transition period, only declarations of conformity to ISO 14971:2019 will be accepted by FDA.
ISO 14971 Relations with other standards
ISO 14971 has relationships with various standards: ISO standards such as ISO 9000 (Quality Management Systems), IEC 62366 (Usability), ISO 13485 (Medical Devices – Quality Management Systems), IEC 60601-1 (electrical medical equipment).
Whereas ISO 60601 addresses single fault, ISO 14971:2019 also addresses a combination of fault modes and hazardous situations as a result of a sequence or combination of independent events. Another example of relation with other standards is the Usability Engineering process (IEC 62366). This process is used for the identification of reasonably foreseeable misuse (in addition to use errors and use associated risks). The outputs of the usability engineering process must be fed back into the risk management process and help complete the identification of hazards. This includes System Security (Cyber Security) and breaches of data. IEC 62304 (Medical Device Software – Software Life Cycle Processes) refers to ISO 14971 for the risk management process of software. ISO 14971 adds an identification of hazards that are related to software that needs to be considered in the process, such as confidentiality, the integrity of data, and availability of data.
ISO 10993-1:2018 Biological evaluation of medical devices requires that the evaluation of overall residual risks associated with the medical device acceptability will be part of the risk management file according to ISO 14971.
It is crucial that traceability will be kept and linked between the various related processes.
An example of this relationship is demonstrated in figure 1.
Figure 1 – Example of relations of ISO 14971 with other standards
ISO 14971:2019 Vs. ISO 14971:2007
ISO 14971 guidance annexes were removed from the standard and are found in ISO TR24971:2020. This new version of the ISO TR 24971 document contains all the normative references and is used to guide the proper implementation of the risk management process.
Revised Terms and Definitions:
New terms that are defined in the standard:
• Benefit: The types of benefits to be considered are discussed: the positive impact of clinical outcome, quality of life, diagnosis, public health. The benefit-risk analysis is aligned to meet MDR and IVDR requirements (the MDR mentions benefit over 60 times vs. 2 times in MDD).
• Reasonably foreseeable misuse: There is an understanding that medical devices can be used for a different intention than the device intended use and that use of the medical device by different populations may result in different outcomes – such as use by medical professionals versus use by laypersons.
• State of the art: This does not necessarily imply the latest most advanced technology. Under the ISO 14971:2019 standard principles, when considering the latest most advanced technology, compared with a more established and widely used technology – it is possible that the benefit-risk perspective of the options will be equivalent. Manufacturers must consider state of the art (clause 10.2), and continually monitor and gather information (generally acknowledged state of the art), and understand if the state of the art changes. This concept is considered in the MDR.
ISO 14971:2019 Scope: The scope of the standard has been clarified to avoid misinterpretation and so specifically mentioning software as a medical device (A.2.1), the Risk Management Process can also be applied to data and security (cyber security), and more detail is given to hazards related to these areas and Radiation, Usability and Biocompatibility. The standard is not limited to Medical Device Manufacturers, but to products that are not necessarily recognized as Medical Device under Regulation and to Suppliers, Contractors, and Service Providers that are involved in the Medical Device life-cycle (compliance with some or all ISO 14971:2019 requirements). • Clause 4.1: The diagram representing the risk management process revised to reflect how the role of the risk management plan in the process. • Clause 4.4: Addition of risk management to include a method for the evaluation of the overall residual risk and requirement to plan criteria of acceptance to this activity (for Medical Device) see clause 8. • Clause 4.5: traceability • Clause 5.4: requires the use of multiple risk analysis tools in order to meet the requirement of identifying known and foreseeable hazards (in both normal and fault conditions) and serve as input to the design process (Annex E ISO TR 24971). This clarifies the need for more than single hazard identification tools: Intended Use; Safety-Related Characteristics; Research/Clinical Trials; Preliminary Hazard-Analysis; Fault Tree Analysis; Usability Engineering Analysis/Human Factors Engineering. During design output, the use of single fault analysis is appropriate in risk management on the design, such as FMEA, SW, and biocompatibility analysis and production. Further identification of hazards is done using data from Risk Control Implementation Verification (Design Verification); Risk Control Verification of Effectiveness (Design Validation phase); Complaints and CAPA process (Post Production) which is also part of the suitability evaluation of risk control measures.
• Clause 8 Disclosure of significant residual risks (Annex A.2.8 ISO 14971 and Annex D ISO TR 24971) Discretion for the analysis of risk/benefit has changed to the requirement to perform a benefit/risk analysis.
• Clause 9 Risk Management Review needs to identify who is going to do the review and when to perform it. Note that the risk management review is part of the risk management fie. The review process can be part of product realization (design reviews).
• Clause 10 Production and Post Production Activities: Expanded and is aligned with clause 8 (Measurement analysis and improvement) in ISO 13485:2016 (and GHTF SG3/N18:2010 QMS MD Guidance on corrective action and preventive action and related QMS processes). Emphasis is given to the active process for gaining information (alignment with EU MDR and FDA Requirements) and inclusion of risk management in post-market surveillance.
• Annex C – Since questions for identification of hazards in the previous editions were taken as mandatory even though, the intention of these questions as guidance was taken out from ISO 14971 and moved to annex A in ISO TR 24971:2020 with additional considerations.
• Annex G – techniques – now annex B in ISO TR 24971:2019 (techniques to support risk analysis). Has additional information to clarify misapplication of techniques, and single-tool use such as FMEA in risk management (see above).
New ISO TR 24971:2020
ISO TR 24971:2013 had some information that did not appear in ISO 14971:2007. The document has been completely revised so it is a very useful guide to risk management and provides guidance with risk analysis, identification of hazards, and evaluation of residual risks – if you follow ISO TR 24971:2020 you can more easily achieve a Medical Device which its’ Risk Management Process conforms with ISO 14971:2019.
Gsap experts will be happy to assist you in updating and preparing your risk management process according to ISO 14971 and related standards.