RNA therapeutics have finally arrived and taken their place as a viable drug discovery platform [1]. Their potential to increase, by orders of magnitude, the number of druggable targets, was evident from the first FDA approval of an RNA therapeutic back in 1998. Only now, however, is this potential being realized with 28 RNA therapeutics now approved globally and in terms of the number of products in development, RNA therapeutics are overtaking unmodified cell therapies [2].
The emergency use and approval of the two mRNA-based vaccines from BioNTech and Moderna shone a spotlight on the potential of RNA-based medicines as a whole, and the speed with which they were manufactured to combat the COVID pandemic gave research towards RNA therapeutics development an immense, and much needed, boost.
In contrast to small molecule drugs and larger biologics, high-quality RNA constructs can be generated faster, and at lower costs; their manufacturing process platform can support any RNA sequence, allowing for personalized RNA therapeutics; and since RNA doesn’t integrate into the host genome, RNA therapeutics have an improved risk/benefit profile.
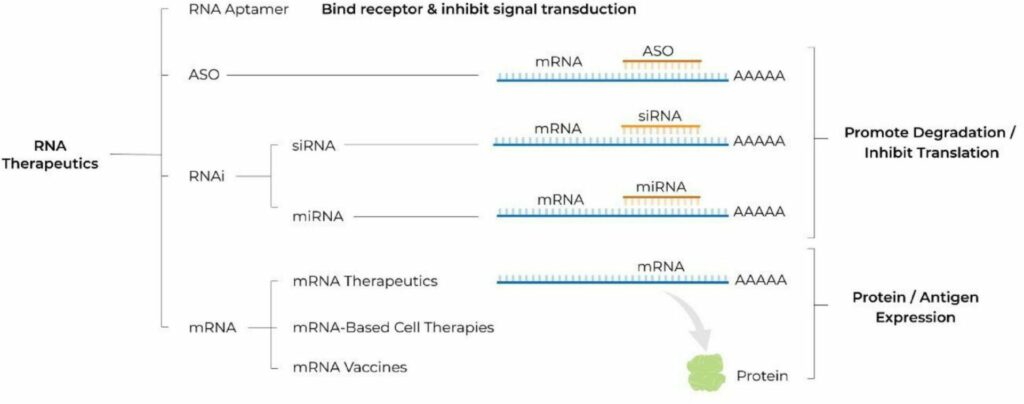
RNA’s intermediary position in the expression of genetic information from DNA to protein presents a huge number of pharmacological targets that were previously undruggable by monoclonal antibodies or small molecules. Moreover, this central position provides a unique versatility to modulate gene expression to introduce new transcripts for protein replacement therapy and more [3]. RNA therapeutics are a diverse group and span from antisense oligonucleotides (ASOs), small interfering RNA (siRNA), microRNA (miRNA), and messenger (mRNA). In general, ASOs and RNA inhibition (RNAi) therapeutics promote RNA degradation and inhibit translation, whereas mRNA therapeutics promote protein or antigen expression. Based on a recent survey of the RNA-based therapy landscape [4], products leveraging RNAi and mRNAs make up the largest portion of the pipeline at 40% and 37% respectively, but the broader pipeline includes oligonucleotide, double-stranded RNA (dsRNA), and micro-RNA (miRNA) products as well.
RNA, in contrast to DNA, is remarkably unstable and is rapidly degraded by RNases which are ubiquitous in the environment; RNAs’ often large size and strong negative charge hinder transport across the cytoplasmic membrane; and exogenous RNA can be highly immunogenic, promoting cell toxicity and impairment of translation into therapeutic proteins. The recent rapid growth of RNA therapeutics has been due to successes in addressing these challenges of stability, delivery, and immunogenicity; including the ability to penetrate the cell membrane and an ability to escape endosomal entrapment once inside the cell. Chemical modifications of RNA facilitated the shift from completely encapsulated RNA nanoparticles to the use of less complex RNA conjugates (e.g., GalNAc). More recent approaches include circular RNA which is stable against exonucleolytic decay, and bioengineered RNA agents produced and folded in living cells also indicate a favorable stability in human cells [5, 6].
Improvement and innovations have accelerated now that RNA Therapeutics are unequivocally feasible, but drug candidates yet need to complete many steps before clinical use, including manufacturing according to Good Manufacturing Practice (GMP) guidelines, pharmacokinetic (PK) / pharmacodynamic (PD) studies, and safety evaluations. Hospital-based RNA therapeutics programs were predicted to be at the forefront of RNA-based drug development, being best positioned to accelerate the translation of transformative therapies from the lab bench to the patient’s bedside [5].
In the US, some RNA-based therapies are regulated as gene therapies, such as those with viral vector delivery systems and mRNA vaccines; these are regulated as biological drugs under a Biologics License Application (BLA). On the other hand, other RNA-based therapies, such as RNAi products are regulated as small molecule drugs under a New Drug Application (NDA) [4]. It’s important to note that in the US these pathways differ in terms of the lengths of marketing exclusivity awarded upon authorization, and the barriers to competition are different for generic compared to biosimilar products.
Gsap’s Advanced Therapies group makes it their business to stay on top of the latest developments in RNA Therapeutics and intends to leverage our extensive regulatory, manufacturing, and development expertise with Advanced Therapies to help clients develop their novel products for marketing approval. Gsap has considerable experience working with the major Israeli hospitals in establishing cGMP-compliant manufacturing capabilities and quality control methods. We’re familiar with navigating unchartered territory. We’re ready and looking forward to guiding clients along a regulatory-compliant critical development path to market.
Agrawal S. RNA Therapeutics Are Stepping Out of the Maze. Trends Mol Med. 2020 Dec;26(12):1061-1064. doi: 10.1016/j.molmed.2020.08.007. Epub 2020 Sep 25.
Data source: Gene, Cell, + RNA Therapy Landscape Report, American Society of Gene & Cell Therapy and Citeline, 2024. https://www.asgct.org/publications/landscape-report
This post is dedicated to young startups and small companies currently facing numerous challenges, more than ever before. From our experience, we have, unfortunately, learned that during crises, there is often a tendency in young companies to neglect regulatory and quality issues. This is done to accelerate product development and meet investor timelines, under the belief that these matters can be addressed later. However, bypassing essential development stages and neglecting coherent documentation may lead to future difficulties, especially during regulatory submission.
Therefore, we have summarized a concise list of things you shouldn’t compromise on now, to ensure minimal investment when seeking regulatory approval later:
1. Intended use and Indications for use:
Define early on the official description that outlines the purpose for which the product is intended, including its medical function, conditions of use, types of diseases or medical conditions it addresses, and the intended user population. This definition is critical for the regulatory process as it determines the regulatory pathway, impacts clinical requirements guides the verification and validation testing of the product.
This task appears relatively simple, but in reality, it is not so. The Intended Use must be defined based on a deep understanding of the market and medical use, alongside regulatory expertise.
Additionally, it is crucial to consider medical reimbursement options, where alignment with both regulatory approval and reimbursement is necessary.
One common and crucial mistake among startup companies is attempting to define the regulatory classification and pathway of their device on their own, despite having no regulatory expertise, to save money. This can be a costly mistake that may result in significant expenses and time delays later when seeking regulatory approval. It is highly advised to invest the money and hire an expert to develop the regulatory strategy for you – defining the product classification, predicate device (for the US), main V&V tests, clinical aspects, and main QMS requirements for your product and of course the main stages for regulatory submission. The regulatory strategy shall be market-specific.
Another common mistake among startup companies or small firms is assuming that upon regulatory approval, they can immediately start selling the product. In reality, there indeed are companies that received FDA or CE approval but failed to penetrate the market because the product lacks medical reimbursement or an attractive sales channel.
2. Device description:
While it may seem self-explanatory, device description is a critical definition that sets the intentions and boundaries of product development. It must correspond with the intended use, and describe how the product will archive it. It’s important to specify what the product does, its features, design specifications, materials used, functional capabilities, operating principles, and any relevant performance characteristics. Additionally, it may include information on indications for use, contraindications, warnings, precautions, and instructions for use to ensure safe and effective use of the device in clinical settings.
The device description is not marketing material but more of an official statement of the company regarding its product. This will be used as the basis for regulatory submissions and labeling.
Marking materials will be based on the intended use and the device description as they are approved by the regulators. Therefore, it should be done carefully, and taking into account both marketing and regulatory aspects.
Unfortunately, we’ve seen more than once a startup that failed to define an accurate device description and as a result faced many inconsistencies and confusion, especially between what is presented to investors or in the marketing materials and what is permissible under regulation.
3. Risk management:
Besides being a mandatory requirement in most regulations, early-stage (during the planning of the design and development) risk management according to ISO 14971 allows for identifying potential hazards. This early identification enables to define the required verification and validation testing (including the necessity and the scope of pre-clinical and clinical trials) and make corrections and improvements to the product design before costly testing and validation begin.
For risk management to be beneficial in an early stage, more than the product’s design, EMC and SW should be considered. We suggested evaluating the risks of the manufacturing process and user-related form the beginning.
Manufacturing Process- when designing a device, the methods of its manufacturing should weigh into the design. Specially, if the product requires any special process or environmental conditions. For example, if you design a sterile finale product that has a built-in buttery, you will need to identify specific risks inherent in the sterilization of a buttery, and consider materials for the proper sterilization and design the product and its packaging to mitigate the identified risks.
In that matter, scaling up the manufacturing process might raise unexpected process risks, especially for products that require special technologies or specific knowledge. When identified early, the manufacturer might be able to avoid the pain of redesigning the product to scale up the manufacturing process.
User-related risks – once the intended user and environment are determined, risks related to user information perception, cognition, and actions should be identified. Then, when designing the product, you should consider possible controls for these risks.
Specifically, medical device reprocessing by the user may raise a lot of concerns and risks. Reprocessing of medical devices should be as simple and straightforward as possible for the user. When designing with such intention, you should be careful to identify who will conduct the reprocessing and what is their technical understanding (medical care professional? technician? layperson?). Equally important is to identify the environment for the reprocessing process. These considerations have a major effect on the required device’s durability, appearance, how its disassembled or connected, and more.
It important to understate that risk management is not a document but a continuous process that spans the entire product’s lifetime. The earlier it is initiated, the more accurate, comprehensive, and ultimately beneficial it will be.
4. Verification and Validation (V&V) planning:
Planning and conducting V&V activities early in the development phase, guided by risk analysis, are essential for regulatory compliance and product quality assurance. But, for young companies and startups, in ultimately important for budget planning.
Planning V&V early and documenting it systematically can help identify connections between processes, requirements, and tests, potentially saving on expensive laboratory trials later. Mapping required V&V tests also anchors budget management due to their high costs.
While planning the V&V tests, it is critical to conduct a thorough study of the applicable regulations, standards, and guidelines for your device.
5. Adherence to proper development stages and documentation:
Documenting milestones in product development, including version differences and actions taken for each version is crucial even for small teams. Clear documentation ensures consistency and facilitates a comprehensive understanding of project progress, particularly when managing tight deadlines and concurrent tasks.
It’s important to note that the above list outlines only the topics that are not recommended to be neglected even in difficult situations. This list is the bare minimum, and additional quality and regulatory requirements will be required depending on the market you intend to target. Of course, ideally, the development of the product should go hand in hand with quality and regulatory requirements.
In the rapidly evolving field of medical technology, achieving optimal usability remains a significant challenge. As we strive to create devices that are both technologically advanced and user-friendly, several key obstacles emerge. Understanding these challenges is crucial for medical device manufacturers and usability engineers to develop effective solutions.
1. Accommodating Diverse User Needs
The medical device landscape is unique in its wide range of users, from highly trained healthcare professionals to patients with varying levels of technical expertise and physical capabilities. This diversity presents a complex challenge:
Healthcare Professionals: Devices must cater to specialists who require advanced functionalities without compromising efficiency.
Patients: Home-use devices need to be simple enough for users who may have limited technical skills or physical limitations.
Assistive Personnel: Often overlooked, this group includes various crucial roles that must be considered in the design process: Hospital Support Staff, Technicians, Home Caregivers, and Emergency Responders. Many of these users might interact with devices in high-stress situations, adding another layer of usability challenges.
Cultural and Linguistic Factors: In a global market, devices must be intuitive across different cultures and languages.
2. Balancing Complexity and Simplicity
Modern medical devices often incorporate sophisticated technologies and multiple functions. The challenge lies in presenting these capabilities in a user-friendly manner:
Feature Overload: Adding too many features can overwhelm users and increase the risk of errors.
Oversimplification: Stripping down functionality to improve usability may limit the device’s effectiveness.
Critical vs. Non-critical Functions: Determining which functions should be easily accessible and which can be nested in menus.
3. Seamless Integration with Existing Workflows
Healthcare environments are complex ecosystems with established procedures. New devices must fit into these existing workflows without causing disruption:
Interoperability: Ensuring new devices can communicate effectively with existing systems.
Training Requirements: Minimizing the learning curve for new devices to avoid workflow interruptions.
Physical Integration: Considering how the device fits physically within the healthcare setting.
Organizational Effects: Understanding and addressing the broader impacts on organizational structure, job roles, and processes. New devices may necessitate changes in staff responsibilities, and departmental interactions, or even create new roles, potentially leading to resistance or requiring careful change management.
4. Regulatory Compliance vs. Innovation
While not mentioned in the original paragraph, this is a significant challenge worth addressing:
Stringent Regulations: Adhering to FDA and other regulatory guidelines can sometimes limit design choices.
Documenting Usability: The need for extensive documentation of the usability engineering process can be resource-intensive.
Balancing Innovation: Ensuring compliance while still pushing the boundaries of technological advancement.
5. Evolving Technology and User Expectations
As technology rapidly advances, user expectations for intuitive interfaces grow:
Keeping Pace: Ensuring medical devices match the usability standards set by consumer electronics.
Future-Proofing: Designing devices that can adapt to future technological advancements.
Generational Differences: Catering to both tech-savvy younger generations and older users who may be less comfortable with technology.
6. Regulatory Compliance vs. Innovation
Stringent Regulations: Adhering to FDA and other regulatory guidelines can sometimes limit design choices.
Documenting Usability: The need for extensive documentation of the usability engineering process can be resource-intensive.
Balancing Innovation: Ensuring compliance while still pushing the boundaries of technological advancement.
Unbiased Summative Validation: Securing an independent, external team to perform summative validation can be challenging. This step is crucial for regulatory compliance and ensuring unbiased usability assessment, but finding qualified teams without conflicts of interest, managing confidentiality, and integrating their feedback into the development process can be complex and time-consuming.
This addition highlights the importance and challenges of unbiased external validation within the broader context of regulatory compliance and innovation.
7. Ensuring Unbiased Usability Validation
External Team Requirements: Identifying and engaging qualified, independent usability experts who have no prior involvement with the device development.
Confidentiality and Intellectual Property: Balancing the need for transparent evaluation with protecting proprietary information and innovations.
Integrating Feedback: Effectively incorporating insights from external validation into the development process, especially when it conflicts with internal assumptions or preferences.
Resource Allocation: Managing the additional time and budget required for thorough external validation without compromising other aspects of development.
Regulatory Alignment: Ensuring that the external validation process meets all regulatory requirements while providing meaningful usability insights.
The medical device landscape is unique in its wide range of users, from highly trained healthcare professionals to patients with varying levels of technical expertise and physical capabilities. This diversity presents a complex challenge:
Healthcare Professionals: Devices must cater to specialists who require advanced functionalities without compromising efficiency.
Patients: Home-use devices need to be simple enough for users who may have limited technical skills or physical limitations.
Assistive Personnel: Often overlooked, this group includes various crucial roles that must be considered in the design process: Hospital Support Staff (e.g., stretcher bearers, porters), Technicians, Home Caregivers, and Emergency Responders. Many of these users might interact with devices in high-stress situations, adding another layer of usability challenges.
Cultural and Linguistic Factors: In a global market, devices must be intuitive across different cultures and languages.
8. Balancing Complexity and Simplicity
Modern medical devices often incorporate sophisticated technologies and multiple functions. The challenge lies in presenting these capabilities in a user-friendly manner:
Feature Overload: Adding too many features can overwhelm users and increase the risk of errors.
Oversimplification: Stripping down functionality to improve usability may limit the device’s effectiveness.
Critical vs. Non-critical Functions: Determining which functions should be easily accessible and which can be nested in menus.
9. Seamless Integration with Existing Workflows
Healthcare environments are complex ecosystems with established procedures. New devices must fit into these existing workflows without causing disruption:
Interoperability: Ensuring new devices can communicate effectively with existing systems.
Training Requirements: Minimizing the learning curve for new devices to avoid workflow interruptions.
Physical Integration: Considering how the device fits physically within the healthcare setting.
10. Regulatory Compliance vs. Innovation
Stringent Regulations: Adhering to FDA and other regulatory guidelines can sometimes limit design choices.
Documenting Usability: The need for extensive documentation of the usability engineering process can be resource-intensive.
Balancing Innovation: Ensuring compliance while still pushing the boundaries of technological advancement.
11. Evolving Technology and User Expectations
As technology rapidly advances, user expectations for intuitive interfaces grow:
Keeping Pace: Ensuring medical devices match the usability standards set by consumer electronics.
Future-Proofing: Designing devices that can adapt to future technological advancements.
Generational Differences: Catering to both tech-savvy younger generations and older users who may be less comfortable with technology.
Conclusion: Overcoming Challenges through Usability Engineering
While the challenges in achieving optimal usability for medical devices are significant, usability engineering and human-centered design approaches offer powerful tools to overcome them. These methodologies provide a structured framework for addressing the complex needs of diverse user groups, balancing functionality with simplicity, and integrating seamlessly into existing workflows.
By employing techniques such as user research, iterative design, and comprehensive usability testing, manufacturers can create devices that not only meet regulatory requirements but truly enhance the user experience. Involving end-users throughout the development process ensures that devices are intuitive and effective in real-world settings.
As the medical device industry continues to evolve, so too must our approaches to usability. By embracing these human-centered design principles, we can create devices that not only meet the technical demands of modern healthcare but also provide a seamless, safe, and satisfying experience for all users. This approach ultimately leads to improved patient outcomes, increased efficiency for healthcare providers, and a more robust, innovative medical device industry.
For more information about our Medical Device services visit:
When it comes to early-stage drug development, Phase 1 clinical trials are pivotal. These trials are designed to assess the safety, tolerability, and pharmacokinetics of new drugs or treatments. A critical decision for sponsors is where to conduct these trials. Here’s why performing Phase 1 clinical trials in dedicated Phase 1 units with a pool of patients offers significant advantages:
1. Expertise and Experience
Phase 1 units are specifically designed for early-phase trials. They are staffed with experts who are highly skilled in managing the complexities and nuances of these studies. From the initial dosing to monitoring adverse effects, their experience ensures meticulous handling of every aspect of the trial, contributing to more reliable and accurate results.
2. Enhanced Safety Monitoring
Safety is paramount in Phase 1 trials, and specialized units are equipped with advanced monitoring systems to track participants’ health in real-time. These facilities often have 24/7 medical staff and immediate access to emergency care, which enhances patient safety and allows for prompt intervention if needed.
3. Streamlined Operations
Dedicated Phase 1 units are optimized for efficiency. They are designed to handle the specific needs of early-phase trials, from patient recruitment to data collection. This specialization reduces the likelihood of operational delays and ensures that the trial progresses smoothly and on schedule.
4. Access to a Pool of Pre-Screened Patients
One of the biggest advantages of Phase 1 units is their access to a pool of pre-screened patients. These facilities often have a database of individuals who have previously expressed interest in participating in clinical trials and have been pre-screened for eligibility. This accelerates the recruitment process, helping to meet enrollment targets more efficiently.
5. Controlled Environment
Phase 1 units provide a controlled environment that minimizes external variables. This is crucial for obtaining clear, unbiased data on how a drug affects participants. The controlled setting helps ensure that the results are due to the drug itself rather than external factors, leading to more accurate and interpretable findings.
6. Regulatory Compliance
Specialized Phase 1 units are well-versed in the regulatory requirements for early-phase trials. They are familiar with the documentation, reporting standards, and ethical considerations needed to comply with regulatory agencies. This expertise reduces the risk of compliance issues and helps ensure that the trial meets all necessary legal and ethical standards.
7. Participant Comfort and Engagement
Phase 1 units are designed with participant comfort in mind. From private rooms to amenities and support services, these facilities prioritize the well-being of participants, which can improve their overall experience and adherence to the trial protocol.
In summary, conducting Phase 1 clinical trials in dedicated units with a pool of patients offers numerous benefits, including specialized expertise, enhanced safety monitoring, streamlined operations, efficient recruitment, a controlled environment, regulatory compliance, and improved participant comfort. For sponsors looking to navigate the complexities of early-phase drug development, these advantages can significantly impact the success and efficiency of their clinical trials.
For more information about our CRO services visit:
Medical devices have the potential to revolutionize healthcare, but this potential is not just in technical prowess but also in intuitive user interaction. The Food and Drug Administration (FDA) in the United States oversees a meticulous regulatory process for usability engineering, fostering the design of medical devices for safe and effective use in real-world environments.
Key Steps in the Regulatory Framework:
User Research and Task Analysis: This foundational stage involves understanding the device’s intended users, their expertise levels, and specific usage environments. Tasks users will perform are meticulously identified to pinpoint potential challenges and use errors.
Use-Related Risk Analysis (URRA): Potential use errors are evaluated to assess associated risks and hazardous situations that affect all potential end users. This stage helps prioritize areas for design improvements.
Deriving Use-Related Design Requirements: The usability engineer identifies user interface elements critical to caregiver and patient safety, deriving design requirements for the development team.
Formative Evaluations: Throughout development, iterative testing with representative users provides real-time feedback for continuous design refinement. The goal is to mitigate use-related risks to acceptable levels before finalizing the device.
Summative Testing: Once the design is finalized, summative testing formally validates its safety and effectiveness, serving as the final checkpoint before regulatory submission.
Benefits of a Robust Usability Engineering Process:
Enhanced Patient Safety: By minimizing use errors, the risk of patient harm is significantly reduced.
Reduced Training and Support Needs: Intuitive devices require less post-market effort in training and technical support.
Elevated User Satisfaction: Well-designed devices foster a more positive user experience for healthcare professionals and patients.
Streamlined Regulatory Approval: A comprehensive usability engineering program can expedite the regulatory approval process.
Conclusion: The regulatory process for usability engineering plays a pivotal role in ensuring the safety and effectiveness of medical devices. By integrating usability considerations from the outset, manufacturers can create devices that are both technologically advanced and user-friendly, ultimately paving the way for improved healthcare outcomes.
For more information about our services to the Medical Device industry:
Incorporating usability in medical device development is a comprehensive, multifaceted process that spans the entire product lifecycle. At its core, it involves three key phases:
Early-stage user research and requirements gathering, where the needs and capabilities of end-users are thoroughly analyzed;
Iterative design and testing, where prototypes are developed and refined based on user feedback and usability evaluations;
Validation and post-market surveillance, ensuring the final product meets usability standards and continues to perform effectively in real-world settings. This holistic approach not only satisfies regulatory requirements but also significantly enhances the safety, efficacy, and user satisfaction of medical devices.
Medical devices are integral to modern healthcare, facilitating diagnosis, treatment, and patient monitoring. However, their effectiveness extends beyond technical capabilities. A critical, often underestimated factor is usability: the ease with which healthcare professionals and patients can interact with these devices.
Impact on Patient Outcomes:
Usability directly influences patient safety and treatment efficacy. Poor usability can lead to:
Increased Error Rates: Complex interfaces may contribute to misdiagnosis or incorrect treatment administration.
Suboptimal Treatment Delivery: Cumbersome devices may not be used to their full potential, hindering treatment effectiveness.
Elevated Costs: Unintuitive devices require more extensive training for healthcare professionals and at times extensive technical support.
Device Rejection: Both caregivers and patients may abandon difficult-to-use devices, leading to poorer health outcomes.
Regulatory Landscape:
Regulatory requirements worldwide mandate that medical devices undergo rigorous design, testing, and monitoring to ensure they are safe, effective, and user-friendly. Integrating human factors throughout the development process, conducting thorough usability testing, and documenting these activities are essential components. Post-market surveillance further monitors device usability in real-world settings to continually improve patient safety and device effectiveness.
The FDA’s Human Factors and Usability Engineering Guidance outlines expectations for incorporating human factors into device design and development. While not legally binding, this guidance reflects the FDA’s current perspective and serves as a valuable resource for manufacturers aiming for regulatory approval. Compliance with FDA recommendations is crucial as industry standards evolve, ensuring devices meet usability expectations and enhance patient care.
Under the EU MDR, similar stringent requirements are binding in Europe. Manufacturers must integrate usability engineering into the entire device lifecycle, perform systematic usability evaluations, and manage usability-related risks effectively.
ISO 62366 helps put these requirements into practice by providing a structured approach to Usability Engineering. The standard guides manufacturers in integrating usability considerations across the device lifecycle, including defining and evaluating user needs and risks.
Adhering to FDA guidance, MDR requirements, and ISO standards not only facilitates regulatory compliance but also demonstrates a commitment to producing safe, effective, and user-friendly medical devices that improve healthcare outcomes globally.
Conclusion:
Usability in medical devices is no longer peripheral; it’s a scientific imperative for ensuring patient safety, treatment efficacy, and regulatory compliance. Prioritizing user-centered design principles can foster improved patient outcomes and streamline regulatory approval processes.
For more information about our Medical Device services visit:
Computer system validation (CSV) is an essential process in the life science industry to ensure that computer systems used in the manufacturing, testing, and distribution of drugs are accurate, reliable, and secure. The guidelines provide a framework for the life cycle of computerized systems validation. The GAMP 5 is a set of guidelines established by the International Society for Pharmaceutical Engineering (ISPE) which is used by pharmaceutical and medical device manufacturers as well as suppliers to ensure compliance with regulatory requirements.
The following are some of the challenges of computer system validation:
▪ It can be a complex and time-consuming process. ▪ It requires a high level of expertise and knowledge. ▪ It can be expensive. ▪ It can be difficult to maintain compliance with regulatory requirements.
Despite the challenges, computer system validation is an essential process for ensuring the quality and safety of products and services.
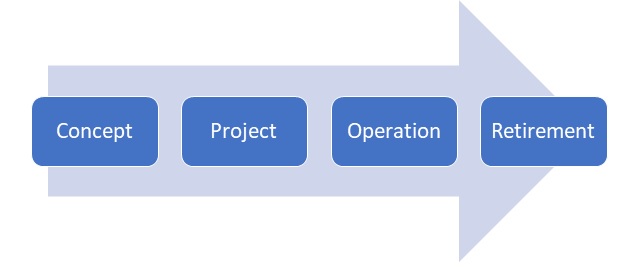
The life cycle of computerized systems validation, consists of four phases which are:
Concept:
▪ Define the system: The system should be defined in detail, including its purpose, scope, users. ▪ Identify risks: The system’s risks should be identified, including if its related to GxP and the questions we had in the priviest post.
Project:
🔸Planning
should cover all required activities, responsibilities, procedures, and timelines, life cycle activities should be scaled according to:
System impact on patient safety, product quality, and data integrity (risk assessment)
System complexity and novelty (architecture and nature of system components, including maturity and level of configuration or customization)
Outcome of supplier assessment (supplier capability)
🔸Specification, Configuration and Risk assessment
Initial user requirements are often gathered during the concept phase and refined during the projected phase. The role of specification is to enable systems to be developed, verified, and maintained. The number and level of detail of the specifications will vary depending upon the type of system and its intended use. For example, software design specifications are not expected from the regulated company for non-custom products.
Specifications should be adequate to support subsequent activities, including risk assessment, further specification and development of the system, verification as appropriate, and system maintenance and update. The requirements for configuration and coding activities depend on the type of system (see Section 4.2.6 for examples).
Any required configuration should be performed in accordance with a controlled and repeatable process. Any required software coding should be performed in accordance with defined standards. The need for code reviews within the organization producing the software should be considered.
Configuration management and version control are intrinsic and vital aspects of controlled configuration and coding
🔸Verification
Verification confirms that specifications have been met. This may involve multiple stages of reviews and testing depending on the type of system, the development method applied, and its use. Verification activities occur throughout the project stages. Testing computerized systems is a fundamental verification activity. Testing is concemed with identifying defects so that they can be corrected, as well as demonstrating that the system meets requirements. The use of effective and appropriate testing tools is encouraged. Such tools should have documented assessments for their adequacy (refer to EU Annex 11 Clause 4.7 [32]) and be controlled in use; however, validation is not required. Testing is often performed at several levels depending on the risk, complexity, and novelty. Tests may be defined in one or more test specifications to cover hardware, software, configuration, and acceptance.
🔸Report and Release
After the verification part of the project, we have the reports that approves that all verifications were completed, and that the system is approved for use. The system should be accepted for use in the operating environment and released into that environment in accordance with a controlled and documented process. Acceptance and release of the system for use in GXP regulated activities should follow a defined process and involve oversight and input of the process owner, system owner, and the appropriate quality function as necessary and applicable. A computerized system validation report should be produced summarizing the activities performed, any deviations from the plan, any outstanding and corrective actions, and provide a statement of fitness for intended use of the system. The incorporation of lessons learned/after action review stage gates in the project should be considered (see /SPE A well-managed system handover from the project team to the process owner, system owner, and operational users is a prerequisite for effectively maintaining compliance of the system during operation. Traceability is recommended to add to the report to ensure that: ▪ Requirements are traceable back to business process needs ▪ Requirements are addressed and traceable to the appropriate functional and design elements in the specifications ▪ Requirements can be traced to the appropriate verification
As well as demonstrating coverage of design and verification, traceability can greatly assist the assessment and management of change. Traceability should be focused on aspects critical to patient safety product quality, and data integrity.
Operation:
🔸Handover Handover is the process for transfer of responsibility of a computerized system from a project team or a service group to the operations team or a new service group. This is an important process; achieving compliance and fitness for intended use on its own may not be enough to guarantee a successful transfer into the operational phase. The handover process will typically involve the project team), process owner, system owner, and Quality. The support group should be involved at the earliest opportunity to ensure effective knowledge transfer and establishment of operational procedures. A period of elevated support and maintenance, often referred to as Hypercare Services, may be arranged to facilitate the transfer. Implied context-specific knowledge acquired through personal experience or internalization, as well as explicit knowledge captured and codified in documentation, tools, and systems should be considered.
🔸Service Management and Performance Monitoring Service management and performance monitoring are shown related to records management due to records generated to demonstrate proper operation and performance of a system. In addition, there is potential interaction with incident and problem management and CAPA and change management when the results of the service or monitoring indicate there are issues that need addressing.
Establishing and Managing Support Services
The support required for each system, and how it will be provided, should be established. Support may be provided by external service providers or internal resources. This process should ensure the following:
▪ Service agreements ▪ Maintenance plans ▪ SOPs ▪ Support systems
2. Performance Monitoring
Performance monitoring detects issues that could impact the availability and performance of the system in order to facilitate mitigation before problems occur. Detected issues are managed through the incident management and problem management processes. Monitoring tools and automation are increasingly used to detect potential issues, report issues, and escalate to support organizations for timely intervention and rectification, and are encouraged.
The need for performance monitoring should be considered, and required activities scheduled and documented. This may change during the operational life of a system.
🔸Incident and Problem Management and CAPA
Incident Management and Problem Management
The incident management process aims to categorize incidents to direct them to the most appropriate resource or complementary process to achieve a timely resolution; whereas problem management involves analyzing root causes and preventing incidents from happening in the future. There should be a procedure defining how problems related to software, hardware, and procedures should be captured, reviewed, prioritized, progressed, escalated, and closed.
This includes the need for processes to monitor progress and provide feedback. Incident and problem management processes may be supported by service management tools that support the incident and problem management process, associated action planning, and traceability of actions taken to address the incident or problem.
🔸Corrective and Preventive Action
CAPA is a process for investigating, understanding, and correcting discrepancies based on root-cause analysis, while attempting to prevent their recurrence. In the operational environment the CAPA process should feed into the overall CAPA system used for GxP operations. When incidents occur, or when opportunities to reduce process/system failures are identified by other means, CAPA should be identified and processes established to ensure that these are implemented effectively.
🔸Change Management
Change Management
Change management is a critical activity that is fundamental to maintaining the proper functioning and controlled status of systems and processes. All changes that are proposed during the operational phase of a computerized system, whether related to software (including middleware), hardware, infrastructure, or use of the system, should be subject to a formal change-control process. This process should ensure that proposed changes are appropriately reviewed to assess impact and risk of implementing the change.
Regression analysis and regression testing may be required. The process should ensure that changes are suitably evaluated, authorized, documented, tested, and approved before implementation, and subsequently closed.
The process should allow the rigor of the approach, including the extent of documentation and verification, to be scaled based on the nature, risk, and complexity of the change, by application of critical thinking. Some activities such as replacements and routine system administration tasks should be covered by appropriate repair or system administration processes.
Change management should provide a mechanism for prompt implementation of continual process and system improvements based on periodic review and evaluation, operational and performance data, and root-cause analysis of failures.
🔸Configuration Management
Configuration management includes those activities necessary to precisely define a computerized system at any point during its life cycle, from the initial steps of development through to retirement.
A configuration item is a component of the system that does not change as a result of the normal operation of the system. Configuration items should only be modified by application of a change management process. Formal procedures should be established to identify, define, and baseline configuration items, and to control and record modifications and releases of configuration items, including updates and patches.
🔸Repair Activity
The repair or replacement of defective computerized system components, which are often but not exclusively hardware or infrastructure related, should be managed in accordance with a defined process. Such activities should be authorized and implemented within the wider context of the change management process. Many repair activities are emergencies and require rapid resolution, so the incident and change management processes should be designed to allow such activities to occur without delay or increased risk to the operational integrity of the computerized system.
🔸Periodic Review
Periodic reviews are used throughout the operational life of systems to verify that they remain compliant with regulatory requirements, fit for intended use, and meet company policies and procedures, including those related to data integrity. The reviews should confirm that, for components of a system, the required support and maintenance processes and expected regulatory controls (plans, procedures, and records) are established.
Periodic reviews should be:
Scheduled at an interval appropriate to the impact and operational history of the system. Risk and other assessments should be used to determine which systems are in scope and the frequency of periodic review.
Performed in accordance with a predefined process
Documented with corrective actions tracked to satisfactory completion
🔸Continuity Management-
Backup and Restore
Processes and procedures should be established to ensure that backup copies of software, records, and data are made, maintained, and retained for a defined period within safe and secure areas
Restore procedures should be established, tested, and the results of that testing documented.
🔸Business Continuity Planning
Business Continuity Planning (BCP) is a series of related activities and processes concerned with ensuring that an organization is fully prepared to respond effectively in the event of failures and disruptions, covering local and global infrastructure, data, and the application
Critical business processes and systems supporting these processes should be identified, and the risks to each assessed. Plans should be established and exercised to ensure the timely and effective resumption of these critical business processes and systems
A business continuity plan defines how the business may continue to function and handle data following failure. It also defines the steps required to restore business processes following a disruption and, where appropriate, how data generated during the disruption should be managed. Plans should be prioritized based on patient and business risk, in case of disruption to multiple systems.
The BCP also identifies the triggers for invoking the business continuity plan, roles and responsibilities, and required communication.
🔸Disaster Recovery Planning
As a subset of BC, plans should be specified, approved, and rehearsed for the recovery of specific systems in the event of a disaster. These plans should detail the precautions taken to minimize the effects of a disaster, allowing the organization to either maintain or quickly resume critical functions. There should be a focus on disaster prevention, e.g., the provision of redundancy for critical systems. Disaster Recovery (DR) plans often require a shared responsibility model between internal organizations of the regulated company and external service providers.
🔸Security and System Administration-
Security Management
Computerized systems and data should be adequately protected against willful or accidental loss, damage, or unauthorized change. Procedures for managing secure access, including adding and removing privileges for authorized users, virus management, password management, and physical security measures should be established before the system is approved for use
Role-based security should be implemented, if possible, to ensure that sensitive data and functions are not compromised. Security management procedures should apply to all users, including administrators, superusers, users, and support staff (including supplier support staff).
Security provisions should ensure that data is protected against unauthorized intrusions including cyber security attacks. Intrusion prevention and detection processes, supported by relevant IT tools and automation, should be in place.
🔸System Administration
Once a system is in operation the users of the system will require support. The system administration process provides administrative support for systems, including performance of standard administration tasks. The extent of this process varies greatly depending on the nature of the system.
🔸Record Management
Retention
Policies for the retention of regulated records should be established, based on a clear understanding of regulatory requirements and existing corporate policies, procedures, standards, and guidelines.
🔸Record Management- Archiving and Retrieval
Archiving is the process of taking records and data off-line by moving them to a different location or system, often protecting them against further changes. Procedures and processes for archiving and effective and accurate retrieval of records should be established based on a clear understanding of regulatory requirements including retention periods.
Retirement:
▪ Retire the system: The system should be retired from use when it is no longer needed. ▪ Archive the data: The system’s data should be archived.
This part covers system withdrawal, system decommissioning, system disposal, and migration of required data.
🔸Withdrawal
Withdrawal is the removal of the system from active operation, i.e., users are deactivated, interfaces disabled. No data should be added to the system from this point forward. Special access should be retained for data reporting, results analysis, and support.
🔸Decommissioning
Decommissioning is a controlled process by which an application or system is removed from use in an organization once it has been retired and/or has become obsolete.
🔸Disposal
Data, documentation, software, or hardware may be permanently destroyed. Each may reach this stage at a different time. Data and documentation may not be disposed of until they have reached the end of the record-retention period, as specified in the company’s record-retention policy, following an authorized and documented process.
Due to the volume of data and records involved, retirement can be a major task for IT systems in particular.
Consideration should be given to:
Establishing procedures covering system retirement, including withdrawal, decommissioning, and disposal as appropriate
Documentary evidence to be retained of actions taken during retirement of the system
GxP records to be maintained, their required retention periods, and which records can be destroyed
The need to migrate records to a new system or archive, and the method of verifying and documenting this process
Ability to retrieve these migrated records on the new system
🔸Data Migration
Data migration may be required when an existing system is replaced by a new system, when an operational system experiences a significant change, or when the scope of use of a system is changed. The migration process should be accurate, complete, and verified.
In conclusion, the GAMP5 guidelines provide a framework for the life cycle of computerized systems validation which is essential in ensuring that computer systems used in the Life Science industry are accurate, reliable, and secure. The five phases of the life cycle, namely planning, risk assessment, specification, testing, and maintenance, provide a structured approach to the validation process, which helps to ensure compliance with regulatory requirements.
Rafael Port Validation & Engineering Project Manager
Are you preparing to bring your medical device to the U.S. market through the 510(k) regulatory pathway? This guide will walk you through the essential steps to ensure a successful submission.
Key Considerations for 510(k) Submission
Define Your Device’s Marketing Strategy Start by clearly articulating your device’s features, benefits, and intended use. This forms the foundation of your marketing strategy in the U.S.
Select an Appropriate Predicate Device Choose a predicate device that demonstrates substantial equivalence based on your marketing claims, intended use, and technical characteristics. This is crucial for establishing your device’s legitimacy.
Determine the Type of 510(k) Submission Identify whether a traditional, special, or abbreviated 510(k) submission is suitable for your device. Tailor your submission accordingly to meet the specific requirements of each type.
Compile Necessary Tests and Data Gather all required tests and data to support your submission. Utilize recognized consensus standards and adhere to relevant FDA guidance to ensure your documentation is comprehensive.
Advocate for Your 510(k) Submission It’s not enough to simply provide evidentiary documents. Advocate for your submission by clearly presenting the data and demonstrating the safety and efficacy of your device.
Maintain Data Integrity and Good Submission Practices Ensure the integrity of your data by following good submission practices. This facilitates a smoother review process and enhances the credibility of your submission.
Familiarize Yourself with eSTAR Starting October 1, 2023, all 510(k) submissions, unless exempted, must be submitted electronically using eSTAR. Understanding this system is essential for compliance.
Understand MDUFA Metrics Be aware of how the Medical Device User Fee Amendments (MDUFA) metrics impact FDA actions and timelines. This knowledge can help in planning and managing expectations.
Engage with the FDA Utilize available options to communicate with the FDA both before and during your submission process. Establishing a dialogue can clarify requirements and expedite the review process.
Know the Role of the CDRH Deputy Ombudsman Familiarize yourself with the function of the CDRH Deputy Ombudsman. This can be a valuable resource if you encounter challenges during the submission process.
By carefully considering these points, you can enhance the likelihood of a successful 510(k) submission, bringing your medical device to the U.S. market efficiently and effectively.
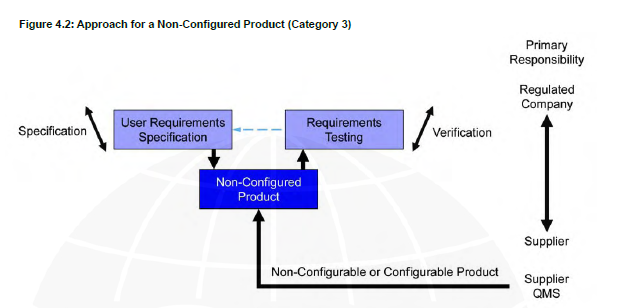
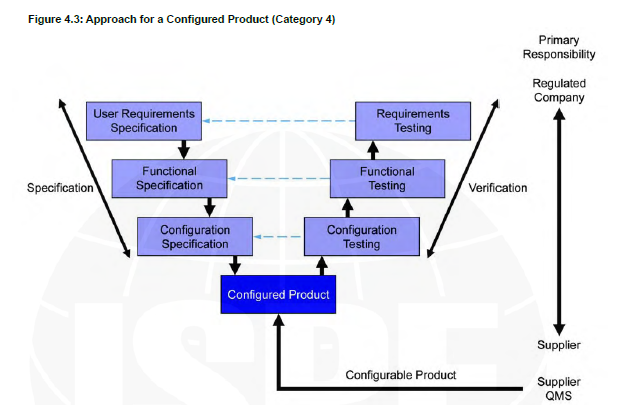
In this post we would like to highlight the relevance of appropriate computer system classification based on GAMP 5 guidelines. GAMP 5 establishes a framework for validating computerized systems in the healthcare sectors. Computer systems should be properly classified in order to determine the amount of risk associated with a system as well as the controls required to reduce those risks. Those actions dictate the validation effort. GAMP 5 divides computer systems into three categories: 3, 4, and 5. (There is another category for software but it’s for Infrastructure Software, Tools, and IT services)
Here is the definition of the different categories:
Category 3
Off-the-Shelf products used for business purposes, which includes systems that cannot be configured and that are configurable but for which only the default configuration is used.
Category 4
Configurable software products that provide standard interfaces and functions that enable configuration of user specific business or manufacturing processes. This typically involves configuring predefined software modules and possibly developing further customized modules.
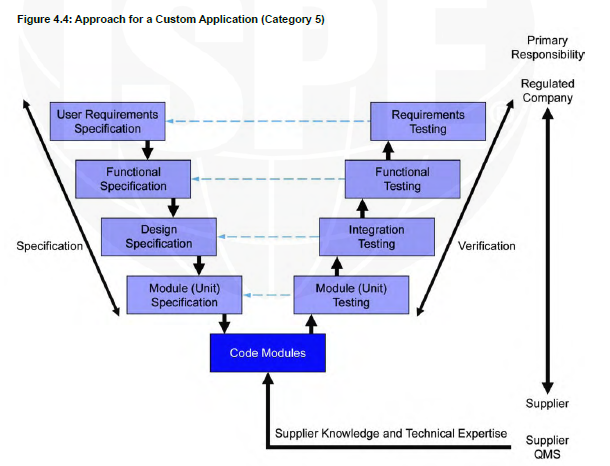
Category 5
Customized systems are developed to meet the specific needs of the users. Custom development may be a completely new system or an extension to an existing system. Complex systems often have layers of software, with one system including components of several software categories; these are commonly treated as Category 5.
V shape model Category 3
For example, UV system that runs on all the spectrumand user’s privileges can’t be changed.
V shape model Category 4
For example, UV system that I can billed different protocol with different light spectrums to fit my needs.
V shape model Category 5
For example, a QMS system that connects to a lab system to collect the date from there and when there is a deviation in the test automatically the QMS will open a deviation form.
As we have seen there is an impact for the different categories on the validation that is need to be performed. So, let’s go to the most important question, How do we determine what category, the system we have or the one we want to purchase? In order to do that we will need first to determine whether this system have any impact on cGxP according to the following bullets:
Automation or control any of: Manufacturing, Sterilization, Formulation, Labeling, Inventory, or Critical Environment Controls
System will be an original source of data for the automation or control of any of: Manufacturing, Sterilization, Formulation, Labeling, Inventory, or Critical Environment Controls
System will use raw and in-process material, clinical data analysis, automated inspection equipment and laboratory data system
System is used to generate, manage and analyze data concerning Product Quality, Safety, Efficacy, Strength Stability or identify
Supporting any GxP Functions such as Calibration, Maintenance Scheduling, and Quality Trending
Manage market complaints or adverse event reporting or electronic document submission/reporting to regulatory agencies
Maintaining copies of protocol pertaining to non-clinical study?
If your answer is No then great, we stop the process here. If you have one answer of Yes, so the system is related to cGxP, and we should move to the next set of questions to decide the systems category.
Was the system developed specifically for the company or any customization done to this application?
If your answer is yes then your system is Category 5 If not we will go to the next question
Is the system a standard product developed by a Vendor where the System-Level Configuration is being modified (excluding Run-Time Configuration) to fit the company’s business process/flow?
If your answer is yes then your system is Category 4 If not we will go to the next question
Is the software a standard product developed by a Vendor and is either
not configurable or
configurable but only the default configuration like run time? (Category 3 – Non-Configured)
If this is the correct then your system is Category 3 If You are not sure contact us for further assistance