The food industry is constantly evolving and we are exposed to the need for new ingredients and products for food and drink. In order to sell food or food products containing components that are not approved for use according to the regulations in Israel, a “Novel Food” approval must be obtained from the National Food Service at the Ministry of Health.
This newsletter will cover the following topics:
- What is Novel Food in Israel?
- Novel Food Regulation
- Genetic Engineered Food
- Labeling of Engineered Food
What is Novel Food in Israel?
Novel Food is defined as food that had not been consumed to a significant degree by humans in Israel before 19 Feb 2006, when the first Regulation on novel food in Israel came into force. ‘Novel Food’ can be newly developed, innovative food, food produced using new technologies and production processes, as well as food which is or has been traditionally eaten outside of Israel.
In order to be categorized as “Novel Food”, the food should belong to one of the following groups:
- Has a new primary structure at the molecular level (e.g. new sugar), which has undergone a deliberate change in the primary structure at the molecular level (e.g. genetically modified food) or originates from a genetically modified organism (GMO).
- Contains or is isolated from plants, animals, microorganisms, fungi or algae (except for enzymes that have a long history of safe consumption in Israel) and does not appear in the lists of edible plants, fungi and algae.
- Has a new production process that results in a change in its nutritional value, metabolism or the level of unwanted substances in food (excluding cleaning and disinfection processes).
Examples of Novel Food that was approved to use in Israel include Docosahexaenoic acid (DHA) and Eicosapentaenoic acid (EPA)- rich oil extracted from Schizochytrium sp. microalga, agricultural products from third countries (chia seeds), food derived from new production processes (UV-treated milk) and 2′-O-Fucosyllactose produced by genetically engineered Escherichia coli bacteria.
Cultured meat is an in-vitro culture of animal cells through tissue engineering. These cultured animal cells are grown in a controlled laboratory environment to mimic specific cuts or parts of meat that would be traditionally sold in the market. In the EU, cultured meat would be regulated by the Novel Food Regulation (EU Regulation No 2015/2283) because “food consisting of, isolated from, or produced from a cell culture or tissue culture from animals, plants, micro-organisms, fungi or algae is considered one of the novel food categories listed in the regulation.” Due to growing trade with the European Union (EU), the Israeli food legislation and standardization system are increasingly harmonized to European standards. Similar to Europe, in Israel, cultured meat will fall under the Novel Food category and will be evaluated under the Novel Food premarket authorization process. To date, the application for cultured meat approval by the National Food Service has not been submitted yet in Israel.
Novel Food Regulation
- Relevant Agency:
The National Food Service at the Ministry of Health is responsible for assuring the safety, quality, and authenticity of food for consumers. This is the regulatory agency responsible for the development of food standards and regulations dealing with foods sold in Israel. The agency is also in charge of imported food licensing.
- Regulations
- Novel Food Directive 2006 (004-08)
As detailed in the 004-08 directive for Novel Food implemented since the beginning of 2006 and published on the National Food Service website, any food that can be categorized as Novel (including genetically modified food) undergoes a thorough evaluation process by the National Food Service prior to its market authorization including aspects related to its safety, nutrition, and consumption of the food in Israel. The underlying principles underpinning Novel Food in Israel are that Novel Foods must be:
- Safe for consumers
- Properly labeled, so as not to mislead consumers
- If novel food is intended to replace another food, it must not differ in a way that the consumption of the Novel Food would be nutritionally disadvantageous for the consumer.
Food that was authorized as Novel food will be listed in the Novel food/food components approved list published on the National Food Service website.
Safety Evaluation of Novel Food
Novel food or components of Novel food (including genetically modified microorganisms) must undergo rigorous safety assessments by a team of experts. It must meet all requirements and tests and be safe to use and consume (case-by-case) prior to its marketing.
Safety evaluation of genetically engineered food is related mainly to possible risk factors affecting human health consuming the food such as:
- An increase in the level of the food’s natural toxins. There are foods that naturally contain a certain amount of toxic substances whose amount naturally does not cause a risk from consuming them. For example, vegetables from the Solanaceae family, such as tomatoes, eggplant, potatoes, etc. In this kind of foods, an evaluation is performed assessing whether following the genetic change, there is an increase in the level of natural toxins.
- The presence of new proteins in foods that may be allergens (substances that cause an allergic reaction in people who are sensitive to these substances). In food that has undergone genetic modification, an evaluation is performed assessing whether the modification changed the existing natural proteins to become allergens or whether new proteins that were added cause allergies in humans.
- The presence of substances in genetically modified food that were not present in previous foods and may affect metabolic processes in the body. In performing a genetically modified food safety assessment, an evaluation is performed assessing whether new substances have been added to the food that were not there before, such as hormones and histamines, which may affect physiological processes in the human body.
An engineered food whose genetic alteration causes an unwanted effect, such as increased toxicity or allergens, is not approved.
To date, the engineered food products that have been independently tested and approved by various food authorities around the world have not been found to have health concerns.
Novel Food Pre-Marketing Authorization Process:
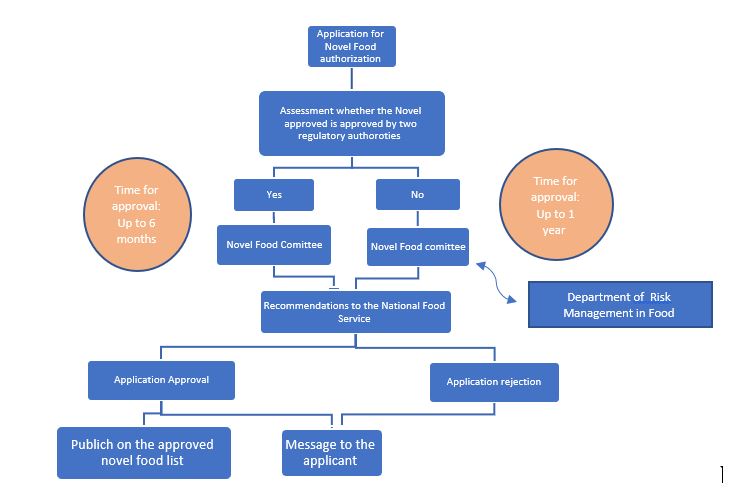
The Public Health (Food) Protection Law – 2015 which came into force in 2016, enshrines the authority of the Ministry of Health to establish safety provisions and regulations regarding the import, sale, and production of Genetically Modified Food (GMO). The main innovation is that the law not only anchors the regulatory authority of novel food safety but also gives the Minister of Health the authority to prescribe provisions regarding its labeling.
In addition to the Public Health Protection Law, there are a number of specific food regulations such as Public Health Regulations (Food) (Gluten Marking), Public Health Regulations (Food) (Marking a Breast Milk Substitute), Public Health Regulations (Food) (Food Additives), Public Health Regulations (Food) (Pesticide Residues), and the Public Health Regulations (Food) (Nutritional Labeling).
- The protection of public health (food) (nutritional labeling) regulations, 5778 – 2017
On December 25, 2017, the Israeli parliament’s Labor, Welfare and Health Committee approved new regulations for the Protection of Public Health (Food- Nutritional Labeling) which entered into force on January 1, 2020. Currently, Israel has no governmental policy on the labeling of Genetically engineered food products (see below).
Genetic Engineered Food
Marketing and labeling of genetically modified food products have caused public turmoil around the world.
The legislative situation in the world and in Israel:
- United States:
There are regulations of various regulatory bodies that deal with food control, such as regulations of the Food and Drug Administration (FDA). At the US there is a requirement to perform a safety assessment for genetically modified food, but there is no special labeling requirement for these products.
- Europe:
In April 2004, Europe entered into force a Regulation (EC 1829/2003) regulating the EU (European Union) guidelines on the safety and labeling of genetically modified food as well as guidelines for approving the release to the environment of genetically modified organisms (GMOs). As part of the installation, there is an obligation to label genetically modified food components or that originate from a genetically modified organism.
- Israel:
In Israel Genetically engineered food is addressed by two committees: The Ministry of Health’s New Food Committee, which discusses food safety, and the Ministry of Agriculture’s Main Committee which discusses Transgenic Plants. Israel is a world leader in agricultural technologies with advanced research on the subject of genetic engineering in plants. Today there is no cultivation of genetically engineered plants for commercial purposes in Israel and therefore there is no local agricultural product that has been genetically engineered. The food industry in Israel does use genetically modified raw materials imported from the USA and other countries, mainly corn, soybeans, and canola oil.

A regulation that legally sets the guidelines regarding Novel Food including genetically modified food and its labeling is in the final stages of legislation. Each novel food before its approval undergoes a risk assessment that includes aspects related to the safety, nutrition, and consumption of the food according to a new food registration procedure that has been implemented since the beginning of 2006 and published on the Food Service website. With the entry into force of new food regulations, the obligation to label genetically modified food components will apply, in addition to a safety examination as has been done to date.
Labeling of Engineered Food
The topic of labeling has become an important topic in discussions about engineered foods all over the world. Government ministries, consumer organizations, and food marketing entities in Israel and around the world believe in the consumer’s right to know whether they have used genetic engineering technology in food production or components from it and therefore require the labeling of these products. In Europe, there is extensive activity on the subject and there are clear requirements for labeling genetically modified food, which are regulated by relevant regulations. In contrast, currently in the U.S. and Canada, it is believed that labeling is not necessary because these foods undergo safety assessment prior to marketing, by a team of experts advising government officials responsible for food control, and only products that are safe to use are authorized for marketing.
In Israel, the Food Service adopts the approach that believes the public has “a right to know” and works to regulate the issue of food labeling that has been legally genetically modified. Once installations will pass, it would create a mandatory labeling requirement for food items that contain genetically engineered ingredients.
Health Vs. functional claims labeling on Novel Food products
In the USA a health claim (i.e a statement about a relationship between food and health) labeled on food products (including Novel Food) is not allowed unless it was approved by the FDA. The functional claim of the food is allowed to be added to the label. The functional claim must be correct but is not approved by the FDA (a disclaimer stating that the claim was not reviewed and approved by the FDA must be shown on the label).
In Europe, there is no differentiation between functional and health claims. Each claim labeled on the food must be correct, established with scientific proof demonstrating the direct relationship between the claim and the food function, and approved by EFSA (European Food Safety Authority).
In Israel, no claim for food is currently allowed (except for phytosterols). Once the Novel Food installations will pass, functional claim labeling for food will be allowed- for Novel Food only.
This Newsletter Prepared by:

Tsufit Gross, Ph.D.
Pharma and Biotechnology Regulation Project Manager