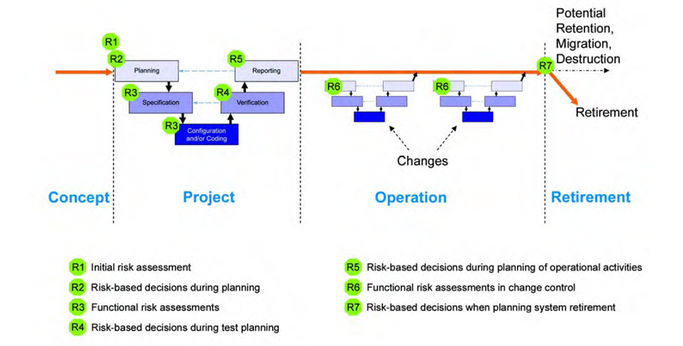
Most companies have a risk methodology in place. In this post, we want to remind you of the critical questions that should guide you during the risk assessment process and the key points where your computerized system can be at risk. But before the questions, do you need to consider your acceptable risk level? The questions we should have in mind: ▪ What can go wrong? ▪ What is the harm? ▪ What is the impact? ▪ What is the probability of a system failure? ▪ What is the detectability of a failure? ▪ How will the risk be managed? The essential thing is to remember that there are risks during the Computerized system life cycle.
If you recognize a risk in one of your systems in one of the different parts of the life cycle, please don’t hesitate to contact us; we would be happy to help you.
The new In Vitro Diagnostic Regulation (IVDR) 2017/746 is a set of regulations issued by the European Union (EU) for the control and supervision of in vitro diagnostic medical devices (IVDs). These regulations aim to improve the safety and effectiveness of IVDs and ensure that they are of high quality.
The IVDR has been applicable since 26 May 2022. In January 2022, the European Parliament and the Council adopted a staggered extension of its transition period, ranging from 26 May 2025 for high-risk in vitro diagnostics to 26 May 2027 for lower risk in vitro diagnostics, and to 26 May 2028 for certain provisions concerning devices manufactured and used in health institutions.
To be prepared for the IVDR, there are several steps that manufacturers, distributors, and other stakeholders can take: Familiarize yourself with the requirements of the IVDR. Make sure you understand the requirements of the IVDR and how they apply to your organization and products. Verify your device classification according to the IVDR new classification system as you may now require the involvement of a Notified Body. Review your current processes and procedures. Ensure that your processes and procedures for developing, manufacturing, and distributing IVDs comply with the requirements of the IVDR. Develop a plan for transitioning to the IVDR, including timelines and budgets. Consider seeking assistance from a third party, such as a regulatory affairs consultancy, to help you navigate the transition to the IVDR. Keep up to date with developments in the IVDR and any changes or updates to the regulations.
By following these steps, you can ensure that your organization is prepared for the IVDR and can continue to provide high-quality IVDs to patients. Contact US for further support and guidance.
This article was prepared by:
Dr. Einat Dekel, DVM
QA & RA Medical Device Senior Consultant, SME In-Vitro Diagnostics & Digital Health
When discussing upgrading the company’s quality system and technical documentation to meet the EU-MDR requirements, many of the companies still think that this is “an activity that QA&RA department should deal with”.
However, the truth is that the main failure cause or bottleneck for upgrading to EU-MDR is the activity related to design and development. For example, product verification and validation tests that have not been performed, design and development documentation that was not kept up-to-date (specifications, drawings etc.), product risk management plans and reports, design changes that were not analyzed and implemented properly and/or were not followed by the relevant verification and/or validation tests, customer complaints that were not analyzed properly as far as influence on product performance and safety is concerned, clinical evaluation documentation including postmarking clinical follow-up and in some cases also the need for clinical trials.
These activities take time, usually, months or even years. Many of these requirements were already there in the MDD, but they were not enforced by the NBs to the extent that they are enforced under the EU-MDR. If the company does not have significant support from the design and development team, the QA&RA departments will not be able to successfully lead the effort to meet the requirements of the EU-MDR. The issues described in relation to design and development, are also critical for successful FDA inspections, which have significantly increased over the last year. It is very important to recognize this fact and understand what needs to be done to achieve compliance.
If you recognize a risk in one of your systems in one of the different parts of the life cycle, please don’t hesitate to contact us; we would be happy to help you.
A traditional clinical trial is carried out at an investigational site, with all patients traveling to and from sites for receiving the investigational treatment and being examined for safety and efficacy. This traditional approach was restricted due to the covid-19 pandemic, and studies around the world were placed in jeopardy. The traditional format of the clinical trials was challenged and led sponsors to look for advanced solutions to overcome and enable clinical operations continuity. Decentralized clinical trials offer a hybrid approach and include solutions that enable more flexibility to patients, clinical staff, and sponsors alike. So, what are decentralized trials, and what advantages do they bring?
A decentralized clinical trial is a modern evolving approach that includes different levels of integration of technological solutions that enable activities and assessments to be performed virtually such as electronic informed consent, video and telemedicine visits, home health, supply chain extensions, and basic remote monitoring. This approach and solutions enable the improvement of data collection flow, recruitment, and patient compliance.
How and why did DCTs become popular?
A major factor for the increase in popularity of decentralized clinical trials was the COVID-19 pandemic. The nature of the global pandemic forced society to find alternative ways to function, and clinical trials were no different. Although decentralized trials were already operating before the COVID-19 pandemic (Pfizer completed its first decentralized clinical trial in 2011), this type of trial became much more popular in response to the global pandemic.
What are the different types of decentralized trials?
Decentralization can be split into two categories:
Fully remote
A fully remote clinical trial is conducted virtually, using mobile devices, remote monitoring software, and apps. Sometimes, home visits are also offered, or patients are required to travel to labs located near their homes. Fully remote clinical trials take advantage of recent advances in telemedicine, and there is no defined “study site” to speak of.
Hybrid
As the name suggests, hybrid clinical trials combine traditional, on-site clinical trials with remote clinical trials. Some activities are conducted remotely, using the technologies described above, while other activities occur at designated study sites. This model is becoming increasingly popular with study sponsors across a wide range of fields and industries.
What are the advantages of decentralized clinical trials?
Decentralization comes with many advantages, including the following:
●Flexibility. Because the model reduces the need to physically travel to a designated study site, a DCT offers much more flexibility for patients. Using telehealth and wearables, data can be submitted from anywhere, at any time.
●Ease of participation. Reducing the need to travel to a study site makes it much easier for patients to participate in clinical trials. The option to participate remotely reduces a major barrier and allows more participants to enroll in a trial.
●Saves time. Recent research has shown that decentralized phase 2 clinical trials can be completed one to three months faster than traditional trials, with phase 3 trials also saving time compared to their traditional counterparts.
●Better generalizability of trial results. With fewer barriers, more patients can enroll in decentralized trials. This makes it easier for trials to enroll a more diverse patient population, which allows for fewer caveats and greater generalizability of the overall results of the trial.
●Improved patient engagement. Patient engagement is higher, and patients are more likely to participate in a clinical trial for the entire duration if the “patient burden” is reduced. The flexibility of decentralization significantly reduces the patient burden, improving patient engagement.
●Authenticity and reliability of data. Due to the use of technology such as wearables and real-time monitoring devices, decentralized trials often lead to more robust data.
●Improves enrollment potential. Transportation is a major barrier to trial enrollment. With decentralized clinical trials, this barrier is largely removed, which improves the potential patient pool.
● Real-time study overview. Collecting data remotely, and in real-time, means that the study team can identify missing data or deviations that must be addressed immediately. Real-time monitoring is especially advantageous for safety data, as any adverse events can be acted upon immediately.
What are the challenges of decentralized clinical trials?
While decentralization offers several advantages, there are also some challenges associated with this newer trial format, including:
●Infrastructure. Clinical trials require a great deal of infrastructure, including financial support, information systems, technology, staff, and more. Coordinating this remotely, and ensuring that all patients receive and administer medications appropriately is a significant logistical challenge.
●Technological skills needed by participants. DCTs often involve patients administering their medication, or affixing their own wearable devices. This requires patients to have some skills in these areas or risks the integrity of trial data.
●Design and planning. DCTs require comprehensive planning to promise smooth execution. The DCT approach requires interaction and interface of many factors and the process needs to be well planned, validated, and documented
●Data quality. Some decentralized clinical trials rely on self-reported data, which may increase certain forms of bias and not be as accurate as data collected from a traditional trial site.
●Patient safety. Patient safety needs to be carefully considered in DCTs. As patients are often not required to report to a trial site, stringent trial monitoring needs to be in place to ensure that all patients are appropriately and frequently assessed for adverse events.
●Regulations. As patients are given more freedom with a DCT, trial staff must ensure that regulations are adhered to despite the reduced patient contact and interaction.
The future of clinical trials
The technological development landscape and regulatory approach towards DCT enable the integration of advanced solutions to improve main parameters in the execution of clinical trials, in particular aspects of recruitment, duration, and cost. Yet, the full integration of applicable solutions should be considered and built in the early stages of the trial design.
Gsap CRO supports sponsors during the planning phase and clinical trial protocol development to promise appropriate integration of the relevant DCT solutions into their operation. This increases the value to the sponsor, improving data authenticity and reducing patient burden.
Clinical trials with Gsap
Gsap is a boutique Contract Research Organization (CRO) with vast experience across a wide range of industries. Gsap’s full-service CRO services will help ensure your trial runs smoothly from start to finish. Whether you are running a traditional clinical trial, a hybrid trial, or a fully-remote clinical trial, Gsap has the professionals on staff to make it a success.
A boutique CRO offering personalized support
The team at Gsap ensures that you have much more than an organization to plan and execute all facets of your clinical trial. Gsap acts as a partner, providing personalized services based on your trial’s specific needs.
Gsap has helped hundreds of clients execute clinical trials across cell and gene therapy, medical cannabis, medical devices, digital health, and beyond. The Gsap holistic approach to CRO services helps to ensure the success of your clinical trial.
Contact Gsap today to discuss your upcoming clinical trial.
This article was prepared by:
Matti Hoggeg, M.Sc.
Clinical Section Manager
For more information about our CRO services visit:
Contract research organizations (CROs) can be excellent assets for companies in the biotech, pharmaceutical, or medical device industries. But what can a CRO actually bring to your company, and when should you enlist the services of a CRO? Let’s take a look at what a contract research organization is, the services offered by CROs, the benefits of CROs, and how to choose a CRO that meets your clinical study’s needs
In short, a contract research organization (CRO) is a company or business that offers support for clinical studies in the biotech, medical device, or biopharma industries. CROs are experts in clinical trial execution, their experts include different disciplines such as CPMs – Clinical Project Managers, CRAs – Clinical Research Associates, Medical Writers, Data Managers etc., thus they can help companies, businesses, or institutions (usually referred to as trial “sponsors”) reduce the costs and often lengthy timelines associated with a clinical study.
What does a contract research organization do?
Now that we’ve addressed what a CRO is, let’s take a look at what a CRO does. A clinical research organization is hired by a clinical trial sponsor and is then responsible for the planning, management, and execution of the entire clinical trial.
The role of a CRO and its assigned team is to ensure that a clinical trial is performed to the highest standards, according to the guidelines and regulations, to ensure the participant’s safety is maintained and that all data is robust and accurate. At the same time, the clinical research organization is responsible for performing the trial efficiently and as cost-effectively as possible.
What services does a clinical research organization provide?
The CRO acts as the main point of contact and coordinates between the sponsor and other stakeholders in the clinical trial, including regulatory bodies, ethics committees, physicians/medical staff, research coordinators, and vendors.
Specific services typically offered by a clinical research organization include:
● Study planning and design ● Medical writing of study protocol and other core documents ● Study feasibility, start-up, and document preparation ● Site selection and activation ● Regulatory submissions and contracts ● Site management and clinical monitoring ● Data management, data analysis and biostatistics ● Clinical trial Project management ● Interim and final study reports ● Quality assurance (QA) and audits
Contract research organizations (CROs) have the skills and knowledge required to run a successful clinical trial and ensure that all elements of the trial are compliant with local and international requirements.
What are the benefits of a CRO?
There are a lot of advantages associated with a contract research organization. The most important include:
Expertise and experience
Most sponsors looking to undertake a clinical trial do not have experience in all of the areas listed above. Therefore, teaming up with a CRO that has the resources, personnel, and experience needed to execute a successful trial offers a much simpler and more streamlined approach to conducting a clinical trial.
Cost-effectiveness
Another major benefit is cost reduction. A clinical research organization that can keep a clinical trial running according to a pre-determined schedule can save the sponsor significant financial burdens associated with delays to the trial.
Meeting the technological needs of the trial
Clinical research organizations have the needed infrastructure and are equipped to meet the clinical and technological requirements of a clinical study, when a sponsor may otherwise not have been able to. This could relate to everything from SOPs, Clinical Trial Management System, subject enrolment tools, to data collection system, biostatistical and data management programs.
Time-saving
If a sponsor was required to create or obtain everything necessary to run a clinical trial, this would take years to commence. A clinical research organization already has all these necessary resources, meaning that a trial can get up and running significantly faster with a CRO for healthcare.
Navigating evolving and complex regulatory requirements
Clinical trial regulatory requirements are ever-changing, complicated to navigate, and often region-specific. A contract research organization can coordinate with regulatory personnel and ensure that necessary approval from the relevant regulatory bodies is obtained prior to the start of the trial. Throughout the trial, a CRO ensures all regulatory requirements are adhered to.
How to choose a Contract Research Organization
Depending on the requirements of a specific clinical trial, a sponsor may have a large number of seemingly suitable contract research organizations to choose from. When assessing which CRO is the best for your specific needs, consider the following factors:
● Is the CRO responsive, communicative, and collaborative? ● Does the CRO have relevant experience? ● Do they have an established quality system and QA processes? ● Is there a high turnover of clinical research associates (CRAs), or is staffing stable? ● What are the services the CRO offers?
These questions, along with others specific to the nature of your clinical trial, will help you find a CRO that gives you the peace of mind that your trial management is in safe hands.
CRO services with Gsap
Gsap is a full-service CRO with significant experience in the design and execution of clinical trials. Whether running an early-phase PK study, or an advanced-stage clinical trial, Gsap can provide the support you need to ensure success each step of the way.
A boutique CRO offering personalized support
Gsap is a boutique CRO, which means that you have more than just an organization to manage your trial – you have a partner you can work with every step of the way. Gsap delivers undivided attention to sponsors, both large and small.
Broad experience across multiple disciplines
Gsap’s CRO services span a diverse array of disciplines, including medical devices and digital health. Gsap also specializes in the rapidly growing field of medical cannabis, as well as cell and gene therapy, and the development of new pharmaceutical drugs and biologics.
End-to-end clinical trial support
Gsap is able to provide services that cover all the stages of a clinical trial. Before your trial commences, Gsap offers study design, planning, regulatory submissions, and site selection services. When a trial is wrapping up, Gsap delivers study reports, closes out study sites, and analyzes the associated trial data and writing interim and final reports.
Customized clinical trial services
By design, no two clinical trials are identical. Therefore, Gsap tailors its clinical trial services to each sponsor, to ensure the needs of your specific trial are met, based on your product’s characteristics and the geographical location(s) of your trial.
Gsap has helped numerous clients successfully manage the transition from pre-clinical development to clinical studies and can support your trial from start to finish. The Gsap holistic approach to CRO services helps to ensure the success of your own clinical trial.
Contact Gsap today to discuss your upcoming clinical trial.
This article was prepared by:
Matti Hoggeg, M.Sc.
Clinical Section Manager
For more information about our CRO services visit:
Learn what is A clinical trial protocol, why it’s important, what the protocol should include, and how to conduct a clinical trial with a CRO This article will address the following topics:
Clinical trials must strictly adhere to detailed protocols, which dictate every aspect of a trial. The clinical trial protocol documents the objectives, study design, methodology, statistical analyses, and general organization of a planned trial. It’s a critical document that helps to ensure that all data collected is robust, and also that the safety of trial participants is guaranteed.
What is a clinical trial protocol?
A clinical trial protocol is a written document that forms the backbone of the clinical trial. The protocol is finalized before any clinical trial activities commence, and must be observed and adhered to throughout the trial. A clinical trial protocol provides answers to many of the critical questions that come with a clinical trial, including:
● Why is the clinical trial being conducted?
● What are the questions that will be examined?
● How will the clinical questions be measured?
● Who can participate in the trial?
● What are the procedures that will be done during the trial?
The clinical trial provides details for physicians, study coordinators, and other staff regarding how to execute the trial. It also describes timelines for the trial, as well as ensuring data integrity and patient safety.
Before the trial commences, the protocol must be submitted for review by an ethics committee and be granted formal approval.
Why is a clinical trial protocol important?
A clinical trial simply can’t be completed without a clinical trial protocol. The protocol essentially acts as a set of quality control guidelines, put in place to protect both the participants in the trial and the integrity of data collected throughout the trial, and to ensure uniformity in the execution across all participating sites.
Specifically, a clinical trial protocol is important because:
● It ensures the safety and health of all participants in the clinical trial
● It details a strict and accurate study plan that all physicians, study investigators, coordinators, and trial staff must follow accordingly
● It ensures consistency in data collection and integrity, meaning that data from multiple different study sites can be compared or compiled
● Approval from ethics committees for the conduct of clinical trial cannot be granted without a robust clinical trial protocol
As you can see, the clinical trial protocol is crucial to the success of the trial itself.
What should a protocol for clinical trials include?
A clinical trial protocol needs to include all the information required to carry out the trial. More specifically, the protocol follows good clinical practice (GCP) guidelines or the closely related ISO 14155 guidelines for clinical trials of medical devices. GCP is an international ethical and scientific quality standard, which dictates how clinical trials should be conducted, and the roles of investigators, review boards, sponsors, monitors, and other stakeholders.
So, now that we know what a clinical trial protocol is, here is an overview of what needs to be included in a clinical trial protocol:
Purpose of the clinical trial
The clinical trial protocol must provide enough background information and rationale to outline the purpose of the trial, and justify why the trial is being conducted. The purpose of the trial will likely depend on the phase of the trial; earlier phase trials are usually to collect safety data, while later phase trials are to examine efficacy.
Trial outcomes and measures
The clinical trial protocol also needs to outline the measures that will define whether or not the trial is deemed successful. The protocol must detail measures that assess the safety and, if relevant, efficacy. Typically, a trial has one or two primary objectives and a few secondary or exploratory objectives.
Study participant criteria
Any given clinical trial has a strict set of guidelines relating to who can and cannot participate in the trial. These inclusion and exclusion criteria depend (among other things) on the disease being studied and the treatment being evaluated. For example, participants in a trial drug efficacy trial will likely need to have the condition or disease that the drug is being evaluated for. Exclusion criteria that could prevent a patient from participating in a trial may include age, medical history, or other medications that the patient is currently taking.
Study design and details
Other elements that need to be included in a protocol for a clinical trial include:
● The length of the trial and the number of participants needed
● The schedule relating to the frequency and duration of clinical visits for participants
● Suitable control measures, and whether or not some participants will be issued a placebo
To summarize, a clinical trial protocol must satisfy GCP guidelines, therefore ensuring both the integrity of the trial and the safety and well-being of trial participants.
Conducting a clinical trial with a CRO
As shown above, a clinical trial protocol needs to be extremely thorough and provide all necessary information to satisfy GCP guidelines and ethical considerations. If a clinical trial protocol is incomplete or insufficient, the trial will likely not receive ethics approval, and therefore cannot proceed. In addition, the trial objective must be carefully considered for the trial to meet the target and succeed.
Given that the clinical trial protocol is such a critical component of the trial itself, and essentially dictates how the trial will proceed, it is often necessary to enlist the services of a professional clinical research organization (CRO). A CRO works closely with the study sponsor to ensure that the clinical trial protocol contains all the necessary details for the trial to be approved.
CROs offer medical writers, clinical experts, and regulatory specialists that can assist in the clinical design and protocol development of your trial.
Clinical trials with Gsap
Gsap is a full-service CRO with years of experience in the design and execution of clinical trials. Gsap offers a team of professionals that can work with you to develop the clinical trial protocol, along with providing any other support your trial needs.
A boutique CRO providing personalized support
As a boutique CRO, Gsap ensures that you have more than an organization to develop your clinical trial protocol and manage your trial – you have a partner to assist you every step of the way.
Gsap has helped many clients create clinical trial protocols and manage the transition from pre-clinical development to clinical studies. The Gsap holistic approach to CRO services helps to ensure the success of your own clinical trial.
Contact Gsap today to discuss your upcoming clinical trial.
This article was prepared by:
Matti Hoggeg, M.Sc.
Clinical Section Manager
For more information about our CRO services visit:
In this article, we will discuss clinical trial monitoring in great detail, including what it is, what processes are involved, and why it is essential for the success of any clinical trial.
Clinical trial monitoring refers to the process of overseeing the progression of a clinical trial. Monitoring in clinical trials involves supervising consent documentation, protocol compliance and adherence, data collection and review, safety data collection and reporting, and much more.
The Importance of Clinical Trial Monitoring
The importance of clinical trial monitoring cannot be overstated. Rigorous monitoring is essential for several reasons, including: To ensure that human subjects are treated ethically and their well-being is protected
● To ensure that all data collection is consistent and accurate, particularly across multiple trial sites
● To ensure that all reported data is complete, accurate, and verifiable, with source documentation
● To ensure that the trial is carried out according to the outlined and approved study protocols and SOPs
Clinical trial monitoring is critical to ensure that accurate, reliable data is collected in an ethical manner throughout the trial process.
The Purpose of Clinical Trial Monitoring
The primary purpose of clinical trial monitoring is to scrutinize and verify the quality of the trial at every stage. Monitoring ensures that a trial is conducted according to the study protocol, with all necessary recording and reporting following the relevant standard operating procedures (SOPs), as well as regulatory requirements and Good Clinical Practices (GCPs).
The Three Types of Clinical Trial Monitoring
Clinical trial monitoring falls into three broad categories.
These include:
On-site
On-site clinical trial monitoring requires in-person assessment, carried out by clinical staff or sponsor personnel, at the trial site. On-site monitoring is the traditional form of clinical trial monitoring and was the most common form of clinical trial monitoring prior to the COVID-19 pandemic.
Remote
Remote monitoring in clinical trials has become more accepted in recent years. Unlike on-site monitoring, monitors do not visit trial sites during remote monitoring in clinical trials. Instead, data review and transfer are done using secure, specially-designed digital platforms.
Centralized
Centralized monitoring in clinical trials is the most appropriate option when there are multiple trial sites. Centralized monitoring arose due to the publication of guidelines relating to risk-based monitoring (RBM), and involves the collation of data from all trial sites to a central location for real-time evaluation.
The Services of Clinical Trial Monitoring
As mentioned, monitoring clinical trial processes involves a diverse number of services. These are some of the key elements of clinical trial monitoring:
Protocol compliance and protocol deviations
Strict adherence to study protocols and SOPs is essential in any clinical trial. The study protocol is written, reviewed, and approved prior to the commencement of the study, and any changes to the protocol, or deviations from the protocol, need to be appropriately documented throughout the trial.
Data collection and missing data
Another clinical trial monitoring service is the oversight of data collection. Clinical trial data must be accurate, complete, and verifiable from source documentation. It’s the role of a clinical trial monitor to ensure that this is the case. In addition, the collection of participant data needs to be performed according to the relevant data protection and privacy laws/guidelines.
ICF and Consent Process Review
Informed consent forms (ICFs) are a critical component of patient enrolment. Part of the process of clinical trial monitoring is to oversee patient enrolment and ensure that informed consent from each subject is appropriately obtained and documented.
Source data review (SDR) vs source data verification (SDV)
SDR and SDV are important elements of risk-based quality management. SDV refers to the process of confirming that reported analytical data accurately reflects the source data collected at the trial site, primarily on the case report form (CRF).
On the other hand, SDR refers to the review of source documents as they related to the clinical trial protocol. Compared to SDV, SDR involves a more strategic approach that focuses intently on the quality of data collection and compliance with regard to the study protocol.
IP/device compliance
An investigational product (IP) is a drug or biological product being used in a clinical trial, while an investigational medical device is typically a physical object (such as a pacemaker). Regardless of whether a trial is assessing a product or a device, compliance is integral to the trial’s success. A clinical trial monitor is responsible for ensuring compliance when it comes to how the investigational product or device is used in relation to the study protocol.
Safety data collection, review, and reporting
Safety data is one of the most important pieces of information gleaned from the study. Many trials are conducted solely to determine the safety of a potential new drug or device. During the study conduct safety data is collected and reviewed on an ongoing basis in order to ensure subject safety and safe flow of information.
Documentation review and collection
Clinical trial monitoring also involves the collection and review of the numerous documents associated with clinical trials. This process may include a review of CRFs, ICFs, and other source documentation such as hospital records and laboratory notes, and the collection of essential documents required to be part of the Trial Master File (TMF), which is the main repository of trial documentation. A critical component of document review is ensuring that all documentation is complete, legible, accurate, version controlled, and contemporaneous.
Equipment, ancillary supply, and logistics
All equipment being used in a clinical trial such as thermometers, centrifuges, etc. must be appropriately calibrated and validated, with associated documentation such as SOPs, logbooks, and training forms. Clinical trials also require a large number of ancillary supplies such as diagnostic and testing materials, and more. All equipment and materials used in or associated with clinical trials must be GCP grade, stored, and used appropriately. Coordinating this is a critical part of clinical trial monitoring.
Clinical Trial Monitoring with Gsap
Without adequate clinical trial monitoring, significant time and money may be wasted on a trial that does not meet the necessary quality standards for successful submission or review. An experienced and trusted contract research organization (CRO) can ensure that all elements of your trial run smoothly.
Gsap is a boutique CRO with vast experience across a wide range of clinical trial therapeutic areas and phases including cell therapy, gene therapy, cannabis, MD, and DH. Contact Gsap to discuss your upcoming clinical trial.
This article was prepared by:
Matti Hoggeg, M.Sc.
Clinical Section Manager
For more information about our CRO services visit:
Just like any project, a clinical trial requires stringent management and control of everything from budgets to timelines and collaborators. Clinical trial project management involves thorough oversight of the clinical study, beginning before the planning phase commences, and continuing through the data analysis and submission period.
Simply put, clinical trial project management describes the steps and processes that an organization puts in place to ensure that a trial is carried out at the highest standard. Clinical trial project management is typically carried out by a clinical research organization (CRO).
A big component of clinical research project management is ensuring the overall quality of a study; that the resulting data is reliable and robust, and that risk management and patient safety are prioritized throughout the trial. Another aspect of clinical research project management is the time management of the study, ensuring that the trial is completed according to the relevant timelines and deadlines.
The Importance of Project Management for a Clinical Trial
Without proper project management, the quality of the data generated from a clinical trial and the progression of the trial itself may not be up to the necessary standard to reach regulatory approval. With so many critical components, like managing partnerships with third-party vendors and suppliers, adhering to timelines, training all necessary staff, controlling all relevant documents, and much more, clinical trial management is essential to the completion of a successful and high-quality clinical trial.
What are the Roles of a Clinical Trial Project Manager?
A clinical trial project manager is a multi-faceted role with many different responsibilities. Some of the primary roles of a trial manager include:
Overall overview and responsibility of study conduct
While various individuals are responsible for specific aspects of a trial, like clinical data collection, quality assurance, site activation, and so on, the clinical trial project manager plays a broader role in the oversight of each of these elements. A crucial component of clinical trial management is coordinating with all specialists and individuals involved in the study to ensure a successful clinical trial.
Study planning and set up
A clinical trial begins long before the first patient sample is collected. Significant study planning and coordination must first be carried out, and extensive document preparation (including study protocols, SOPs, ICFs, and more) is undertaken prior to the commencement of the trial. Study planning plays a large role in clinical trial project management, and it is critical to work with a clinical trial manager who is able to understand the needs and objectives of the trial, in order to ensure timely progression and compliance of the study. Comprehensive planning for the study start will pay off during the study conduct and save time and budget.
Vendors selection
In a majority of cases, a clinical trial can not be executed by a single study team. Third-party vendors are often included in clinical trials to offer expertise regarding specific technologies or specialized clinical procedures associated with patient sample collection. Typically, there are multiple qualified vendors to choose from. Selecting and vetting the most appropriate vendor(s) is another role that falls under clinical research project management.
Milestones follow up
During the planning phase of a clinical trial, milestones should be established to outline various steps and stages throughout the trial. Milestones may include protocol finalization, regulatory and ethics submission deadlines, study site activation, first subject in/out, last subject in/out, completion of data review and statistical analysis, and site closeouts. The clinical trial project manager is responsible for following up on each milestone as the trial progresses, to ensure that all deadlines are met and the study progresses according to the initial study plan.
Communication and escalation
Effective communication is an essential skill that is required by all clinical trial project managers. Communication with stakeholders, within study teams, between sites, with third-party vendors, qualified personnel, and also to regulators and auditors is critical to the progression and success of the clinical trial. The project manager must also understand when to escalate any issues to the appropriate personnel. The clinical project manager is responsible for internal and external communication as well as action items follow-up, milestones etc. The Communication processes enable the Sponsor and stakeholder to be on the same page and progress the project vectors to the needed direction at each time point.
Collaboration with all parties and vendors
As well as initially selecting vendors, the project manager must maintain a collaborative relationship with vendors throughout the study. Successful clinical project management involves close communication and cooperation with all vendors and third-party players. When it comes time to submit final regulatory documentation, the project manager must coordinate with all stakeholders and vendors to ensure the appropriate materials and documents are obtained and submitted.
Timeline and budget oversight
Clinical trials are inherently expensive, and poor project management can lead to study delays, which further increase the costs associated with the trial. Clinical research project management is primarily focused on adhering to timelines and budgets, with the overarching goal being to ensure that the trial is completed according to schedule, and within the outlined budgets.
Study team management, interphases, guidance, and training
Coordinating, guiding, and managing the study team (including clinical research associates, statisticians, QA personnel, medical advisors, and site staff) is another key role of the project manager. Ensuring that appropriate personnel is fully trained according to study protocols and SOPs (and that training is comprehensively documented) is essential in clinical project management.
Project Management Plans
The project manager is responsible to ensure that each component of the study has a plan in place which is comprehensive, study-specific, current throughout the study, and updated as needed in order to cover all study aspects. The plans include a Clinical management plan (CMP) that outlines the scope and study-specific procedures, a Quality plan that ensures QC and QA procedures will be performed throughout the trial, a Data management plan (DMP) which outlines the plan for the collection and management of clinical data throughout the duration of the trial and more as needed.
Risk management
Assessing and managing risks is a significant element of clinical trial management. Risks might include IRB approval delays, issues with patient recruitment, unexpected staff turnover, unplanned protocol changes, etc. The role of the clinical project manager is to assess the probability of a given risk occurring, the impact that it would potentially have on the trial, and, of course, how to mitigate each outlined risk.
Clinical Research Project Management with Gsap
Gsap is a boutique CRO with vast experience managing a diverse range of clinical trials, including in the pharma, Medical Device, Digital Health, Diagnostics, cannabis, and gene/cell therapy fields. Gsap’s vastly experienced team can manage your project in close collaboration and partnership with all relevant parties and vendors, with respect to timelines, milestones, training materials, sites support, in-house monitoring, open action follow-up, eTMF, CMP, and internal SOPs. This approach ensures a high level of project management and quality of study conduct, leading to the success of your trial. To learn more about Gsap’s clinical trial project management services, contact Gsap here.
This article was prepared by:
Matti Hoggeg, M.Sc.
Clinical Section Manager
For more information about our CRO services visit:
Clinical trials require extensive experience, planning, and execution. With major investments, both in terms of finances and time, it’s crucial to ensure that every step is executed according to plan, from initial protocol development to data analysis. With Gsap, you can be sure that your trial will be carried out to the highest standard, and in compliance with all relevant regulatory requirements.
What are Clinical Trial Services?
Clinical trial services is a broad term that can pertain to any number of outsources services provided during the clinical trial process. Clinical trial services can include everything from the initial protocol development and study planning, to trial execution and data management, and the final data analysis and reporting.
The highly regulated nature of clinical trials, and the necessity to justify every decision made throughout the process, means that outsourcing clinical trial services can bring major benefits. A CRO (clinical research organization) can manage every aspect of the trial process and has the necessary network of suitable sites, professionals, and infrastructure.
Clinical trials are essential for the advancement of safe, effective treatments across all fields of medicine. Whenever a new treatment is developed, such as a drug, vaccine, or medical device, clinical trials must be performed to first assess safety, then efficacy. Drug dosing, contraindications, and additional crucial data can also be gleaned from clinical trials.
While in vitro and animal studies can play crucial roles in early-stage medical research, such as the identification of potential new therapeutic drug compounds, there is simply no substitution for clinical trials. Trials in human subjects, under strictly regulated and controlled conditions, are essential in the evaluation of a new drug, treatment, or medical device.
The Value of End-to-End Clinical Trial Services
End-to-end clinical trial services, or full-service clinical trial management, involves utilizing the expertise of a CRO for the entire clinical trial process. This brings invaluable consistency to your clinical trial, saves significant time in document preparation, and ensures you can work with a trusted partner who understands your goals and priorities.
Common Types of Clinical Trial Services
There are many elements of a clinical trial, all of which must come together to produce the final product. The main types of clinical trial services include:
Clinical trial design and planning
Trial design and planning is arguably the most important element of a successful clinical trial. By enlisting a CRO to assist with establishing the framework for your trial, you bring invaluable expertise from professionals who understand the objectives of your trial and can ensure your study timelines are adhered to.
In the planning and design phase alone, you can save significant time by utilizing a CRO to provide clinical trial services. A CRO can use its extensive network to coordinate everything from study timelines to site selection, ensuring your trial is set up for success from the very beginning.
Clinical trial regulatory services
The regulatory element is a key component of clinical trial services. Thorough documentation, before, during, and after the trial, is essential. Clinical trial regulatory services typically include the preparation of study protocols, which dictate every step of the clinical trial process and include the objectives.
Other necessary documentation that is usually covered in clinical trial regulatory services includes the investigator’s brochure (IB), and informed consent forms (ICFs), which provide a thorough analysis of the potential risks and benefits of clinical trial participation.
A CRO can aid in the management of multiple regulatory submissions, as well as preparing all documentation for ethics committees and other regulatory bodies, such as government agencies (MOH, FDA, etc). Outsourcing such regulatory tasks to CRO specialists results in faster trial start-up and follow-through times, which in turn can lead to significant financial savings.
Medical writings
Clinical trial study reports and associated documentation is a major component of the clinical trial process. Furthermore, there are highly specific guidelines relating to how all documentation must be written and submitted. A CRO that offers specialized medical writing services can save you significant time, and also reduces the risk of delays resulting from missing or incorrect documentation.
Clinical trial management
Overseeing a clinical trial, both in terms of the day-to-day processes and adherence to all protocols and SOPs, and the broader study progression relative to the required timelines is a monumental task. Trial management is a key clinical trial service provided by CROs, and involves everything from site selection to qualification of the study team. Budget oversight is also a critical component of clinical trial management.
Clinical trial data management
Data management for a clinical trial begins long before the trial itself. Planning what data will be collected, at which time points, and how it will be collected, stored, and interpreted, is an essential part of the clinical operation. Data management is a key clinical trial service offered by CROs, and gives you the peace of mind that all trial data is collected in line with both the clinical protocol and relevant regulatory guidelines.
Clinical trial monitoring
Clinical trial monitoring involves rigorous oversight of the study, including documentation, data collection, addressing protocol deviations, oversight of the study team, on site and remote visits, staff training, and more. Enlisting a CRO for clinical trial monitoring services helps to alleviate logistical challenges, keep trial timelines on track, and helps to ensure a smooth path to data cleaning.
Quality assurance (QA)
Another critical element of a clinical trial is quality assurance (QA). QA is responsible for the thorough, unbiased examination of all trial-related documentation. This includes the analysis of SOPs, plans, and protocols prior to the commencement of the trial, as well as all batch records and data collected during the trial.
A rigorous and experienced QA team is essential to ensure that the data collected in a clinical trial, and the manner in which it is collected, meet the necessarily stringent regulatory guidelines.
Clinical Trial Services with Gsap
Gsap is a boutique CRO that offers a wide range of clinical trial services, delivered to the highest standard. With hundreds of satisfied clients ranging from small-scale startups to large international companies, Gsap specializes in trial management across different disciplines, including MD, DH, medical cannabis research, as well as cell and gene therapy.
For end-to-end clinical trial services that can help ensure the success of your trial, contact Gsap.
This article was prepared by:
Matti Hoggeg, M.Sc.
Clinical Section Manager
For more information about our CRO services visit: