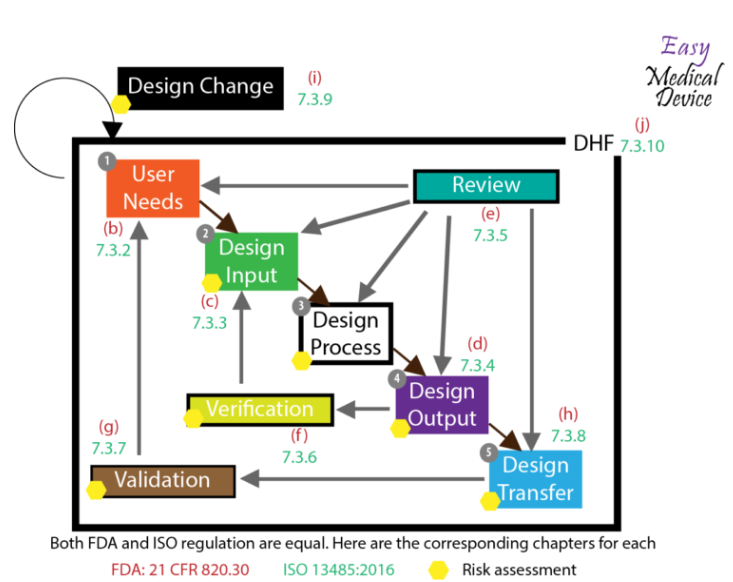
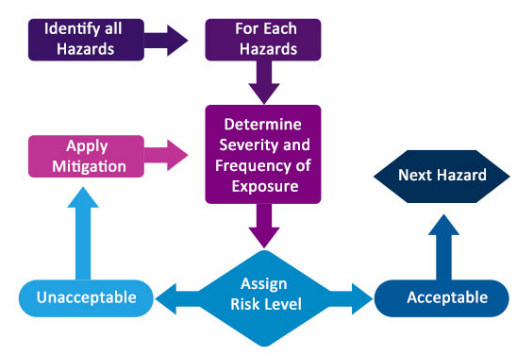
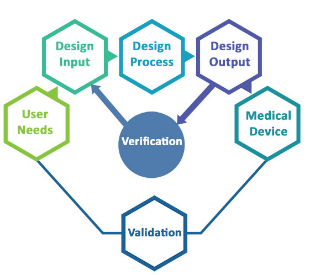
Do you work with software that manages important or even crucial processes in your organization? Are you sure that the results these programs deliver are correct and consistent?
Validation of computerized systems is required for all GXP processes. It is also a useful tool for other fields to make sure the software works well and your information is safe.
What is the conventional risk-based approach to computerized system validation?
The GAMP5 is currently the most widely used industry guidance. It advises businesses to concentrate on critical aspects of their computerized systems and use them to develop controls to reduce the risk of system failure. This is where a thorough understanding of the product and process is critical in determining the potential risks to individual safety.
Essentially, the guidance focuses on understanding the system and the process in which it is involved, allowing us to identify the critical functions that impact patient safety, product quality, and data integrity. They are commonly referred to as GxP-relevant functions. Following the implementation and verification of relevant appropriate controls to address the risks, we either redesign the system to eliminate the issues or verify the functionality to demonstrate that the risks can be managed at an acceptable level through execution IQ, OQ, and PQ testing.
What additional risks and regulations can be addressed?
A computerized system frequently plays such a critical business role that we would like it to be verified not only within GxP-relevant processes but also within other realms of critical business processes.
Specific functionality may, for example, generate a financial report to be submitted to public agencies, provide data to a Financial Consolidation System, or support other financial activities that are not necessarily regulated by the relevant regulation (SOX).
Alternatively, specific functionality is used to create, manage, or host personal information about individuals (GDPR), or to manage legal or contract data or transactions.
A system function can be critical for the business in general, like impacting the operations in a way that may lead to stopping the production, shipping, or invoicing process or even lead to legal issues.
How to approach it?
In such cases, to reduce the overall effort for commissioning the system, we will evaluate the criticality of the requirement in a manner similar to GxP evaluation, and if the function is critical for a specific business process, we will manage it as if it were a GxP critical function.
Figure 1: GAMP5 A Risk-Based Approach to Compliant GxP Computerized Systems, Appendix M3
According to GAMP5, before evaluating the Risk Class, we must first determine the Severity, which is defined as “Impact on Patient Safety, Product Quality, and Data Integrity.” If you’re implementing a system in your organization that manages other critical processes (especially ERP systems), you might want to broaden the definition of “Severity” to include additional questions, which will result in a system with reliable performance with the critical parts defined by the business.
If your company relies on systems whose failure could halt critical processes, alter the final product, or incur enormous costs, you should take extra precautions to ensure the system consistently produces the desired results and that there is objective evidence to support this claim.
The risk-based validation paradigm widely used within Pharma and Medical Device environments can be simply adapted for use in other fields.
This article was prepared by:
Vitaly Shlimovich
Engineering & validation project manager
For more information about our Validation services visit:
Creating a medical device requires you to concentrate on the process of documenting and prototyping phases. In highly regulated areas such as healthcare, a quality management system (QMS) is crucial to the development of a new product.
The question is what is the minimum QMS for the design and development stage? To answer this question, we recommend considering the following:
A medical device should be safe and effective to reduce product recalls and revisions, demonstrate compliance with all relevant regulatory standards, and be commercialized quickly.
General recommendations for QMS needed at the development stage for medical device products:
● Documentation should demonstrate consistency, reproducibility, and repeatability. Furthermore, documentation should communicate what to do, when to do it, and how to do it effectively.
●You should ‘right-size’ your QMS as you build it. In other words, the size of the QMS should reflect the size of your organization. By ‘right-sizing’ your QMS, you ensure that you build the quality procedures you need at the right time. The needs of a 5-employee company differ from one of 50 employees and from 500 employees.
●You need to make sure you consider the product realization and the lifecycle of the device. Identify all the documents that you will be required to generate as part of these processes.
●Be sure to determine who is the owner of each document, for how long it must be kept, and where it will be stored before you begin documenting anything.
●Consider the availability of documentation and an effective and efficient way of managing the documentation. Medical device companies are strong when they manage their documentation well.
We recommend using an Electronic Quality Management System (eQMS). If you choose eQMS, you need to define your documentation and record control with your eQMS.
●Developing and updating your document management strategy takes a different approach as your company becomes more established. It is advisable to break down your document management system into phases and plan each phase carefully.
The Bare minimum required QMS documents:
(1) Regulatory Strategy Document
The regulatory strategy includes product description, product intended use and indication for use, product classification, regulatory pathway per relevant markets, applicable product verification and validation tests, list of applicable standards and guidance
(2) Design and Development Control Procedure
The design and development control procedure describes the method for initiating, controlling, and documenting product design processes.
It is recommended that the procedure address the following processes:
●Processes for defining the user needs, product value proposition, and VOCs (Voice of Customer),
●Processes for defining product specification (design inputs),
●Risk Management processes in accordance with ISO 14971:2019 including failure mode analysis
●Guidelines for defining Sample size and rationale for design validation and process validation processes,
●Processes for design verification and design validation
Purchasing control procedure includes processes to facilitate supplier management, guidelines to oversee the purchase of materials, components, products, or services according to quality standards, and conformance to all specified requirements. These shall include receiving inspection processes and control of purchasing records.
(4) Document and Record Control
This is one of the most important procedures for the design and development phase. Many manufacturers are not aware of how critical it is to document the design at the very early stages of design and development. This includes a methodology for numbering and traceability of all documentation and records that are created from the very beginning of the design and development (such as design inputs, design outputs, test protocols, any test results, reports, design reviews, and any changes to the design, risk management file).
Document and record control procedure means defining a formal document control process from numbering documents and records to adding/maintaining/changing/removing documents and records.
(5) Engineering Change Orders (ECOs)
ECO is an important tool that ensures that changes that are done to a product(s) under development are identified, reviewed, validated/verified, documented, and approved before their implementation. It is highly recommended to define the ECO process for stages before design freeze (or design output review) and after design freeze. Changes before the design freeze shall include as a minimum: Details of the change, the reason for the change, and the approver of the change. All records must be kept in all phases.
(6) Quality Manual
A full quality manual is not required at the early stages of the design and development, yet a frame with the building bloc needs to be defined for the quality manual. This frame needs to include the scope of the quality management system and its exclusions, a list of the planned documented procedures for the quality management system, and a cross-reference table to later include the relationships between the QMS procedures and the regulatory requirements.
It is also advisable to include in the quality manual the company’s organizational structure, specifying the functions in the organization.
Conclusion
At the beginning of the development of a new product, it is necessary to plan for the evolution, including the minimum documentation required at this step, thinking about the right size and procedures of the QMS.
Targeted cancer therapy continues to evolve, and many new treatment combinations are being approved in addition to developmental advancements in novel checkpoint inhibitors. This article will address the lately approved antibody therapeutics (US and EU), those in regulatory review, the expanded approval of existing immunotherapies, and their associated companion diagnostics for cancer treatment. This article will address the following issues:
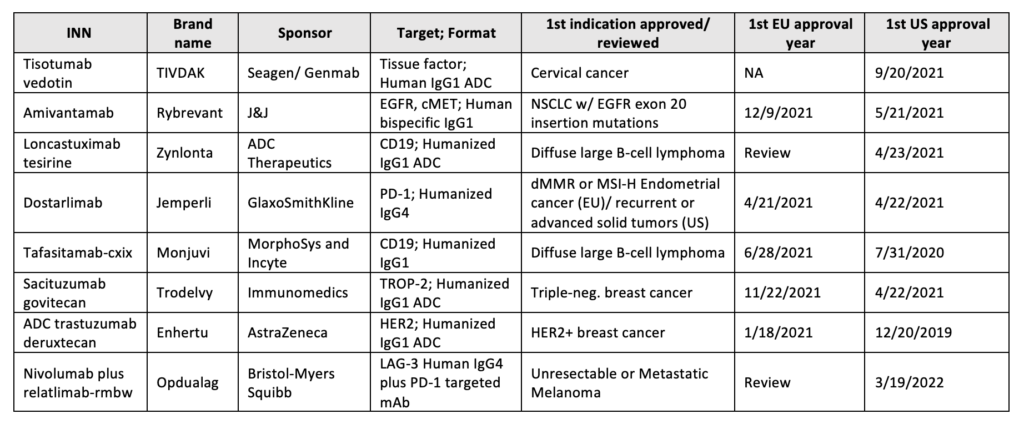
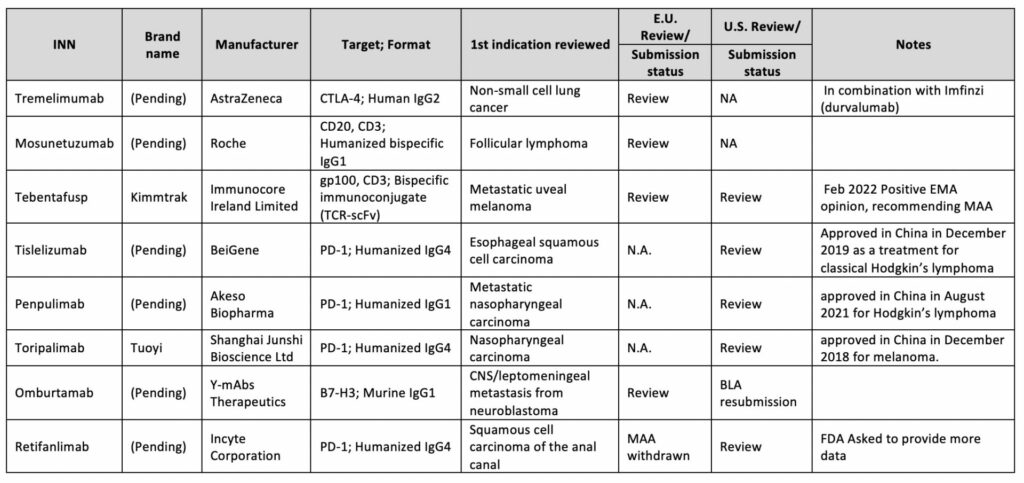
Between 2021 to 2022, the FDA approved six new antibody-based immunotherapies and EMA five whereas four of the five accepted were previously cleared by the FDA, Table 12,3.
Table 1: Antibody therapeutics approved in the EU or US
Starring 2021’s approval is Dostarlimab (Jemperli), anti-PD-1 Humanized IgG4, and Amivantamab (Rybrevant), a bispecific antibody targeting cancer epitopes, EGFR and MET. Both were approved in the EU (M.A. No. EU/1/21/1538/001) and US (BLA:761174/761210) to treat tumors with a high mutational burden and NSCLC with EGFR exon 20 mutations, respectively.
Companion Diagnostics and Future Artificial Intelligence (AI)-Based Approaches
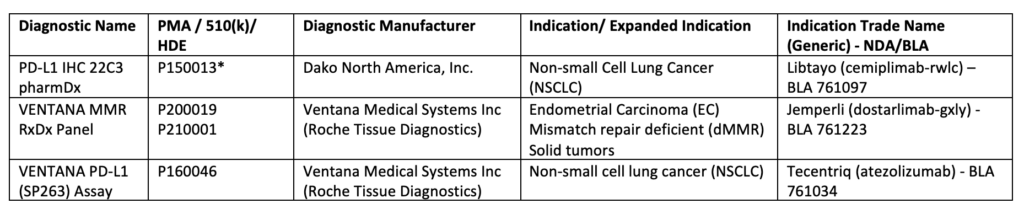
The use of immune signature to predict immunotherapeutic response led to an increase in the development and approval of immunohistochemical (IHC) diagnostic assays, Table 24. Such devices allow informative decision-making to assess tumor-favoring immune responsiveness5,6. For example, the VENTANA MMR RxDx panel was approved concomitantly with Dostarlimab (mentioned above) to distinguish the patients with dMMR who will benefit from this treatment7. Additional FDA-approved companion diagnostic tests are listed below, Table 2.
Table 2:List of Approved Companion Diagnostic Devices
Important past year’s (2021) step in the immunotherapeutic approach is the expanded indication approvals for several already in-use drugs as adjuvant/neo-adjuvants therapies for the core axis PD-1/PD-L1. PD-1/PD-L1 blockade was proven to promote DFS (Disease-Free Survival) and EFS (Event-Free Survival). Atezolizumab (Tecentriq) had been cleared by the FDA as an adjuvant treatment with the companion diagnostic test VENTANA PD-L1 (SP263), mentioned above, for NSCLC (non-small-cell lung cancer) patients expressing PD-L113. Pembrolizumab (Kytruda) was approved both by EMA and FDA for the adjuvant treatment of RCC (Renal Cell Carcinoma) and neoadjuvant+adjuvant treatment for TNBC (Triple Negative Breast Cancer), respectively 14,15 . Nivolumab(Opdivo) has also been approved by the E.C. (European Commission) and the FDA for the adjuvant treatment of esophageal or GEJ (gastroesophageal junction) cancer16,17. The latter had also approved Nivolumab for adjuvant therapy of urothelial carcinoma18.
Latest Advancements in the Immunotherapy Field and 2022 Expected Approvals
From a regulatory point of view, the unexpected immune response of antibody-based biopharmaceuticals is one of the obstacles when developing biological products. Thus it is advised to navigate the strategic and regulatory decisions correctly to properly assess the safety and efficacy of such biotherapeutic 22. These assessments are usually conducted during pivotal clinical trials; as per ICH S6(R1), nonclinical immunogenicity data are not considered to be predictive of clinical safety 23. Even a greater challenge arises when designing antibody-based immunotherapy with expansion to dual targets i,e. Bispecific Antibodies (bsAbs). Such a strategy may increase immunogenicity issues which may also be related to novel complex structures. Consequently, for benefit-risk assessment, the FDA may request a comparison of the bsAb to an approved monospecific product directed against the same antigenic target(s)24.
In any circumstance, every applied BLA needs to show a clinical advantage. For example, FDA denied the approval of Retifanlimab (PD1-targeted mAb)25and Oportuzumab monatoxalso (EpCAM-targeted immunotoxin, ADC), questioning their clinical benefit. In addition, concerns were raised about the Oportuzumab monatoxalso company’s CMC (Chemistry, Manufacturing, and Controls) processes26.
Accelerated approvals are granted to many I.O. biologics; however, some are not reviewed for years. In 2021, FDA conducted an industry-wide evaluation of such drug approvals, which failed to meet post-marketing requirements 27,28. Following FDA’s call into question, several immunotherapy indications were withdrawn and removed from the US market28,29,30.
1. Cox EM, Edmund A V, Kratz E, Lockwood SH, Shankar A. Regulatory Affairs 101 : Introduction to Expedited Regulatory Pathways. 2020;2012:451-461. doi:10.1111/cts.12745
2. Www.antibodysociety.org.17Dec2021. Antibody therapeutics approved or in regulatory review in the EU or US. Antib Soc. Published online 2021. https://www.antibodysociety.org/resources/approved-antibodies/
3. Mullard A. 2021 FDA approvals. Nat Rev Drug Discov. 2022;(January). doi:10.1038/d41573-022-00001-9
4. List of Cleared or Approved Companion Diagnostic Devices (In Vitro and Imaging Tools). doi:https://www.fda.gov/medical-devices/in-vitro-diagnostics/list-cleared-or-approved-companion-diagnostic-devices-in-vitro-and-imaging-tools
5. Emily A. Prince, PharmD1 ; Jenine K. Sanzari, PhD1 ; Dimple Pandya, MD1 ; David Huron P; and RE. Analytical Concordance of PD-L1 Assays Utilizing Antibodies From FDA-Approved Diagnostics in Advanced Cancers : A Systematic Literature Review abstract. Published online 2022. doi:10.1200/PO.20.00412
6. Christian U. Blank, John B. Haanen AR and TN. The cancer immunogram. Science (80- ). 2016;352(6286):658-660. doi:10.1126/science.aaf2834
7. VENTANA MMR RxDx Panel – P200019. Published 2021. https://www.fda.gov/medical-devices/recently-approved-devices/ventana-mmr-rxdx-panel-p200019
8. Baxi V, Edwards R, Montalto M, Saha S. Digital pathology and artificial intelligence in translational medicine and clinical practice. Mod Pathol. 2022;35(1):23-32. doi:10.1038/s41379-021-00919-2
9. Xu Z, Wang X, Zeng S, Ren X, Yan Y, Gong Z. Applying artificial intelligence for cancer immunotherapy. Acta Pharm Sin B. 2021;11(11):3393-3405. doi:10.1016/j.apsb.2021.02.007
10. Artificial Intelligence and Machine Learning in Software as a Medical Device_Action Plan. https://www.fda.gov/medical-devices/software-medical-device-samd/artificial-intelligence-and-machine-learning-software-medical-device
11. Good Machine Learning Practice for Medical Device Development: Guiding Principles. https://www.fda.gov/medical-devices/software-medical-device-samd/good-machine-learning-practice-medical-device-development-guiding-principles
12. Roche announces the release of its newest artificial intelligence (AI) based digital pathology algorithms to aid pathologists in evaluation of breast cancer markers, Ki-67, ER ,and PR. https://diagnostics.roche.com/global/en/news-listing/2021/roche-announces-release-of-its-newest-ai-based-digital-pathology-algorithms-to-aid-pathologists-in-evaluation-breast-cancer-markers-ki67-er-pr.html
13. FDA F and DA. FDA approves atezolizumab as an adjuvant treatment for non-small cell lung cancer. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-atezolizumab-adjuvant-treatment-non-small-cell-lung-cancer
14. FDA F and DA. FDA approves pembrolizumab for high-risk early-stage triple-negative breast cancer. Published 2021. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-pembrolizumab-high-risk-early-stage-triple-negative-breast-cancer
15. EMA. Committee for Medicinal Products for Human Use (CHMP) adopted a positive opinion for the extension of Kytruda as monotherapy adjuvant treatment_RCC. Published 2021. https://www.ema.europa.eu/en/medicines/human/summaries-opinion/keytruda-6
16. Bristol Myers Squibb. Bristol Myers Squibb Receives European Commission Approval for Opdivo (nivolumab) as Adjuvant Treatment for Esophageal or Gastroesophageal Junction Cancer Patients with Residual Pathologic Disease Following Chemoradiotherapy. Published 2021. https://news.bms.com/news/corporate-financial/2021/Bristol-Myers-Squibb-Receives-European-Commission-Approval-for-Opdivo-nivolumab-as-Adjuvant-Treatment-for-Esophageal-or-Gastroesophageal-Junction-Cancer-Patients-with-Residual-Pathologic-Disease-Following
17. Bristol Myers Squibb. U.S. Food and Drug Administration Accepts for Priority Review Application for Opdivo® (nivolumab) as Adjuvant Therapy for Patients with Resected Esophageal or Gastroesophageal Junction Cancer. Published 2021. https://news.bms.com/news/details/2021/U.S.-Food-and-Drug-Administration-Accepts-for-Priority-Review-Application-for-Opdivo-nivolumab-as-Adjuvant-Therapy-for-Patients-with-Resected-Esophageal-or-Gastroesophageal-Junction-Cancer/default.aspx
18. FDA F and DA. FDA approves nivolumab for adjuvant treatment of urothelial carcinoma. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-nivolumab-adjuvant-treatment-urothelial-carcinoma
19. Kaplon H, Reichert JM. Antibodies to watch in 2021. MAbs. 2021;13(1). doi:10.1080/19420862.2020.1860476
20. Si Y, Melkonian AL, Curry KC, et al. Monoclonal antibody-based cancer therapies. Chinese J Chem Eng. 2021;30:301-307. doi:10.1016/j.cjche.2020.11.009
21. Vaddepally RK, Kharel P, Pandey R, Garje R. Review of Indications of FDA-Approved Immune Checkpoint Inhibitors per NCCN Guidelines with the Level of Evidence. :1-19.
22. Vandivort TC, Horton DB, Johnson SB. Regulatory and strategic considerations for addressing immunogenicity and related responses in biopharmaceutical development programs. J Clin Transl Sci. 2020;4(6):547-555. doi:10.1017/cts.2020.493
23. European Medicine Agency. EMA/CHMP/ICH/731268/1998 ICH guideline S6 (R1) on preclinical safety evaluation of biotechnology-derived pharmaceuticals. Comm Med Prod Hum use ICH. 2011;6(June):1-22. https://www.ema.europa.eu/en/documents/scientific-guideline/ich-s6r1-preclinical-safety-evaluation-biotechnology-derived-pharmaceuticals-step-5_en.pdf
24. Gough J, Nettleton D. Bispecific Antibody Development Programs – Guidance for Industry_CEDER. Manag Doc Maz. 2019;(April):10.
25. Tucker N. FDA Denies Approval of Retifanlimab for Locally Advanced or Metastatic SCAC Subgroup. Published 2021. https://www.targetedonc.com/view/fda-denies-approval-of-retifanlimab-for-locally-advanced-or-metastatic-scac-subgroup
26. Biospace. FDA Slams Sesen Bio with CRL for Bladder Cancer Drug. Published 2021. https://www.biospace.com/article/fda-crushes-sesen-bio-with-crl-for-bladder-cancer-drug/
27. Astor L. FDA Cracks Down on Dangling Accelerated Approvals in 2021, Pathway Is Scrutinized. Published 2021. https://www.targetedonc.com/view/fda-cracks-down-on-dangling-accelerated-approvals-in-2021-pathway-is-scrutinized
28. FDA In Brief: FDA Oncologic Drugs Advisory Committee to Review Status of Six Indications Granted Accelerated Approval. Published online 2021. https://www.fda.gov/news-events/fda-brief/fda-brief-fda-oncologic-drugs-advisory-committee-review-status-six-indications-granted-accelerated
29. Merck withdraws Keytruda from SCLC indication amid FDA crackdown. Published 2021. https://www.clinicaltrialsarena.com/news/merck-withdraws-keytruda-for-lung-cancer-amid-fda-crackdown/
30. Nivolumab Indication in Small Cell Lung Cancer Withdrawn in U.S. Market. Published 2021. https://ascopost.com/issues/january-25-2021/nivolumab-indication-in-small-cell-lung-cancer-withdrawn-in-us-market/
This article was prepared by:
Vered Ben Hur. Ph.D
Pharma Regulation Projects Manager
For more information about our Pharmaceuticals industry visit:
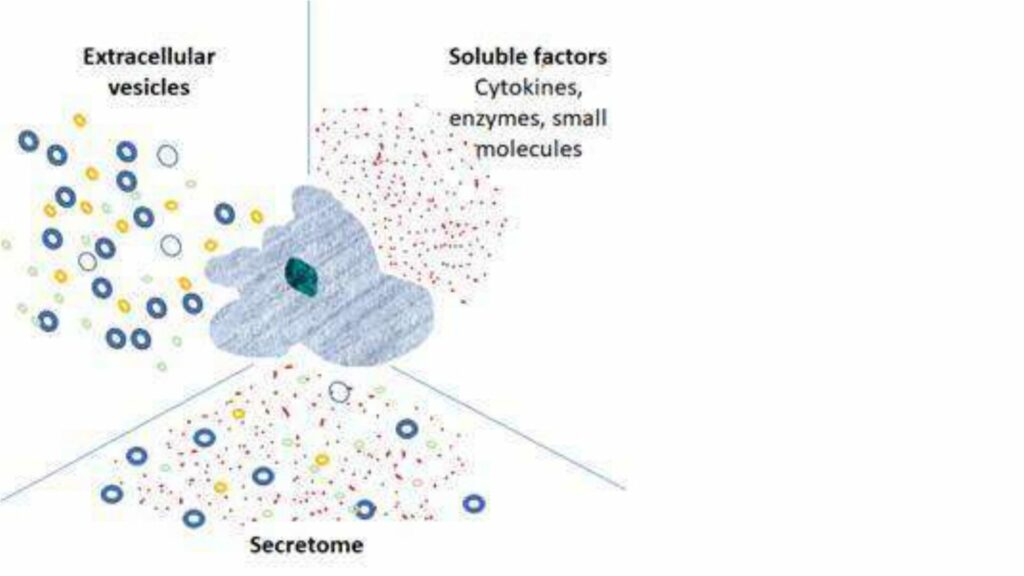
In the last decade, the cell therapy field has matured, with some notable successes, including CAR-T therapies and gene-modified cell therapies targeting specific rare diseases. There have also been some disappointments, in particular, the failure of several MSC therapies to reach efficacy endpoints in late-stage clinical trials. In parallel, however, the realization that very many beneficial effects of cell therapy can be attributed to the cells’ secretome, has gained traction. This is evident in the exponential increase in publications relating to the secretome and extracellular vesicles (EVs), as well as in the number of start-up companies engaged in the development of such cell-free cell-derived products.
Enter the new kids on the block: cell-derived, cell-free products.
These products are derived from cell culture’s secretions – the secretome – which includes: soluble proteins and peptides, cytokines, chemokines, lipids, carbohydrates, lipid bi-layer bound vesicles including micro-vesicles, extracellular-vesicles (EVs), small EVs (sometimes referred to as exosomes) and their contents, which in addition to the above, contain nucleic acids (DNA and miRNA). The composition of the secretome depends on the cell type and microenvironmental stimuli. This is a dynamic and rich soup!
In addition, to a cell’s natural secretions, companies are engineering and/or activating cells to specifically influence the secretome by, for example, enriching for secretion of a particular factor, or directing expression of a particular factor to the surface of an EV.
What are the advantages of EV’s and Secretome – cell-free cell-derived products?
Cell-free cell-derived products may offer a number of advantages over cell therapies:
In terms of manufacturing – such a product is likely to be more stable, possibly amenable to lyophilization ,and may not require cryopreservation. This represents a huge benefit in terms of process timing, testing ,and transport logistics compared to cell therapies.
In terms of safety – a cell-free product will have no persisting cells and therefore, no potential for transformation and risk of tumor formation. Similar to traditional biological products, an EV or secretome-based product is expected to have a relatively short-term effect with half-life and clearance by predictable pathways.
In terms of efficacy – a cell-free product containing multiple active components enables a broad-spectrum approach to multiple targets, amenable for a range of indications. Repeat dosing is more likely to be required, but dosing should be easier to tailor to the patient’s requirements, based on traditional PK understandings.
What are the main hurdles to be overcome when developing such a product?
One of a kind
All cell-free cell-derived products we’ve encountered at Gsap have been very different from each other in terms of the manufacturing process, product description ,and indication. As with cell therapy, each product is unique with the manufacturing process and controls developed to suit each individual product, and careful risk assessment, is a must, to accompany each step in the development.
Cell banking
Of particular importance is the use of a well-characterized, fully tested cell bank, suitable for GMP manufacture. The cell bank is subject to the same rigorous testing requirements as required for cell therapy. The use of well-characterized commercially available cell banks, manufactured according to GMP, with a well-documented history and testing certification can be of great benefit to enable efficient translation of cell-free cell-derived products into the clinic.
Manufacturing process
Similar to cell therapy, the concept that the product is the process certainly applies here. The scientific literature is replete with examples illustrating the impact of the environment, culturing conditions, cell source, cell type, donor age, media, reagents, scale-, and vessel type, amongst other things, on cell secretome content. Early implementation of appropriate process controls is, therefore, essential to develop a process for a product sensitive to so many variables. EV or secretome-based products may be less potent than cell therapy products unless the manufacturing process can successfully concentrate the main active components whilst eliminating impurities.
No benchmarks
To date, there are no approved EV or secretome-based products approved for marketing in any territory. In contrast, the main risks from cell therapy have been established, over almost two decades of clinical use. In the absence of any benchmark products and potentially more risks, it is critical to generate an understanding for the development product’s mechanism of action (MOA) in order to successfully mitigate potential risks.
Regulatory considerations
Combined regulations are expected to apply to cell-free cell-derived products such as EVs and secrotome products, both in the US and in Europe. The exact product classification may vary, based on the product composition and attributes.
Traditional biological product regulations are expected to apply with regard to nonclinical safety testing, in other words, unlike cell therapy requirements, PK and safe pharmacology evaluation may be required and GLP safety studies may be required in two species. Safety and toxicology programs should be agreed with the regulator in advance. With cell-derived products, the potential for confounding immune response should be carefully considered.
Conclusions
These are exciting times, as biotech companies explore the use of cell secretomes for therapeutic benefit. Such cell-free cell-derived products present a whole new list of challenges for the developer and regulator alike. The new products in development are complex mixtures that require sophisticated analytical methods for their characterization – proteomics, nanoparticle tracking analysis, next-generation sequencing ,and mass spectrometry are just a few of the methods that we can expect to see more of. As with cell therapy, we trust that the individual regulatory bodies will harmonize their requirements so that development is not needlessly complicated further.
This article was prepared by both:
Diana Gershtein MSc., MBA
Advanced Therapies Section Manager
Tami Horovitz, PhD
Gsap Regulatory Submissions – Content Expert
For more information about our Pharmaceuticals industry visit:
As many of you know, the European Union is going through a significant change in the regulation of medical devices. First was the MDD to MDR, and now is the time for the IVDD to transition into the IVDR. This transition has a significant effect on many of the IVD products that will have to go through the new certification process.
‘In vitro, diagnostic medical device’ means any medical device which is a reagent, reagent product, calibrator, control material, kit, instrument, apparatus, piece of equipment, software or system, whether used alone or in combination, intended by the manufacturer to be used in vitro for the examination of specimens, including blood and tissue donations, derived from the human body, solely or principally to provide information on one or more of the following:
(a) concerning a physiological or pathological process or state; (b) concerning congenital physical or mental impairments; (c) concerning the predisposition to a medical condition or a disease; (d) to determine the safety and compatibility with potential recipients; (e) to predict treatment response or reactions; (f) to define or monitoring therapeutic measures. Specimen receptacles shall also be deemed to be in vitro diagnostic medical devices; [Article 2 2017/746]
In simple words, IVDs are medical devices and accessories used to perform tests on samples, such as blood, urine, tissue is taken away from the human body to help detect infection, diagnose a medical condition, prevent disease, or monitor drug therapies.
Some examples of IVDs include systems to test the level of coagulation, urine test strips, home use pregnancy tests, HIV or Hepatitis tests, blood glucose monitoring, etc. IVD device may be used by laypeople (home-use blood glucose level) or by a health professional (detection of N. meningitidis in CSF or blood)
How are IVDs different from medical devices?
An IVD is a subset of a medical device. The legislator created this subcategory and made different requirements for the IVD due to the different risk sets associated with them: no direct contact with the patient, the value of the medical data they deliver, and that they don’t provide treatment.
IVDs fulfill their role based on information they provide, without direct contact with the patient or direct action on the patient. This is of fundamental importance and significantly impacts product validation and testing.
The quality of the information delivered by an IVD is assessed by measuring the analytical precision of the test or assay and the clinical evidence of the information provided. It is evident how important it is for the IVD product to be very precise, for example, when testing blood for blood-typing (identifying blood type) or screening for HIV, COVID-19, etc. We also understand the clinical relevance of these assays – this is why those IVDs with the highest risk potential on the population must have exact performance criteria and results.
The European Union (EU) is beginning a new era
In Europe, the field of IVD has been controlled by the IVD Directive (IVDD) 98/79/EC that was issued in 1998, and compliance became mandatory in Dec 2003. This era is coming to an end as we are shifting to the new generation of the IVD Regulation (IVDR) 2017/746 published on April 5th, 2017, which is currently beginning to come into effect.
The old directive was relatively descriptive, while the new regulation is very prescriptive. To illustrate, the IVDD is 37 pages, whereas the IVDR is 204 pages. The IVDR was written in the same spirit as the MDR regulation, and they have a lot of similarities.
Some of the main points in the IVDR:
●Risk-based classification
●Classification categories were changed.
●Product whole lifecycle
●New subcategories of IVDs (Devices for near-patient testing, Companion diagnostic, Software devices)
●Greater scrutiny of Notified Bodies
●No “grandfathering” provisions. All currently approved IVD devices must be recertified under the new requirements and demonstrate that they are state-of-the-art.
●More rigorous clinical evidence. Manufacturers need to conduct clinical performance studies and provide evidence of safety and performance according to a device’s assigned risk class.
●Requirements for post-market surveillance were significantly increased, and the general timeline for reporting was reduced.
●General Safety and Performance Requirements (GSPR) replaced the “essential requirements.”
●Identification of ‘person responsible for regulatory compliance.’ (PRRC)
●Implementation of unique device identification (UDI) for better traceability and recall (See UDI newsletter in Gsap website)
●More responsibility for economic operators, etc.
Timelines
The IVDR 2017/746 entered into force in May 2017, with a final Date of Application of May 2022 for new devices, with a progressive rollout for devices with a valid IVDD certificate.
However, all IVD products on the market after 26 May 2022 must comply with the IVDR for post-market surveillance, market surveillance, vigilance, registration of economic operators, and devices. [2017/746 article 113]
When talking about the progressive rollout, one must understand some key definitions:
Placing on the Market; [2017/746 (21)] means to supply the product to a distributor or the end-user, not the importer.
Putting into Service [2017/746 (22)] means the first time the product is used commercially and not in a study.
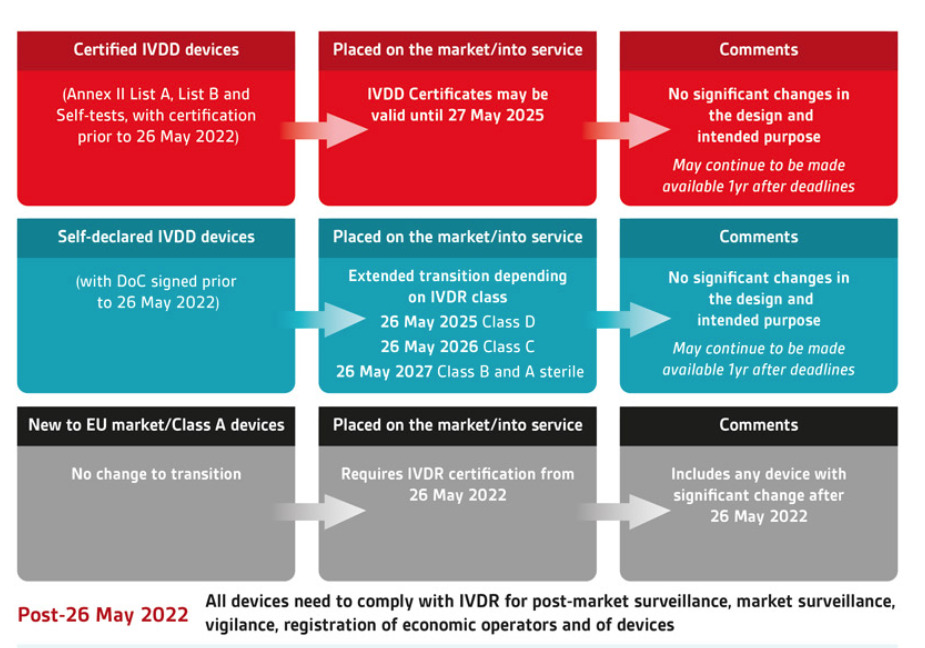
The picture above illustrates how the old and new classifications, together with the definition mentioned above (placing on the market, putting into service), will affect the deadline to:
●When new products cannot be introduced into the market
●When inventory cannot be sold from the manufacturer/ importer to the distributor
●When inventory cannot be used
as you can see, the best-case products that are self-certified and are class A or B under the new classification of the IVDR, can have their products used up to May 2028.
Classification of IVD:
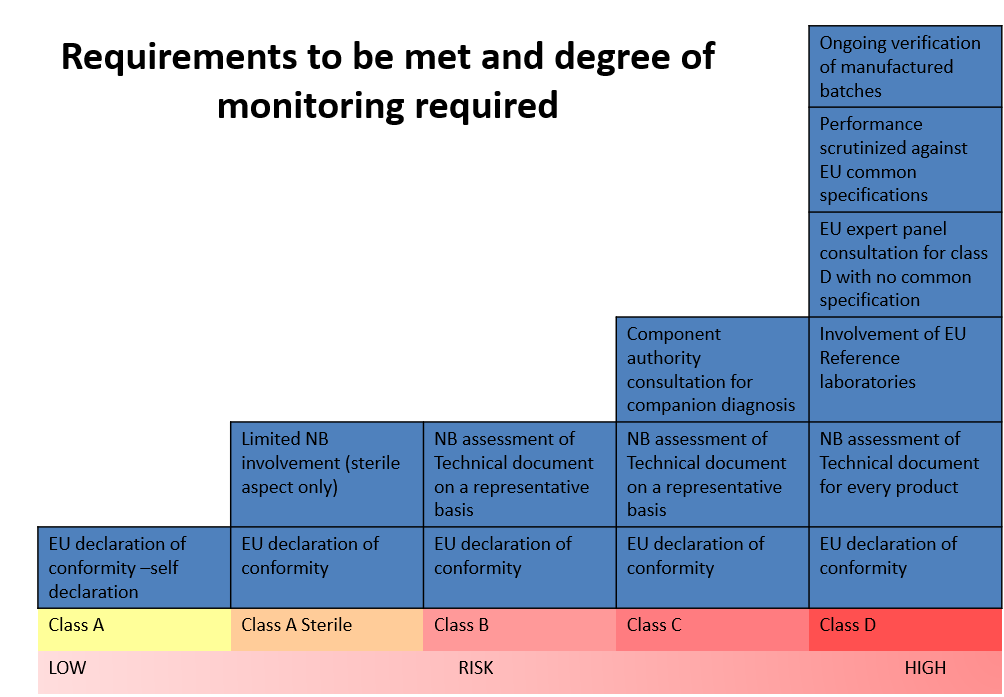
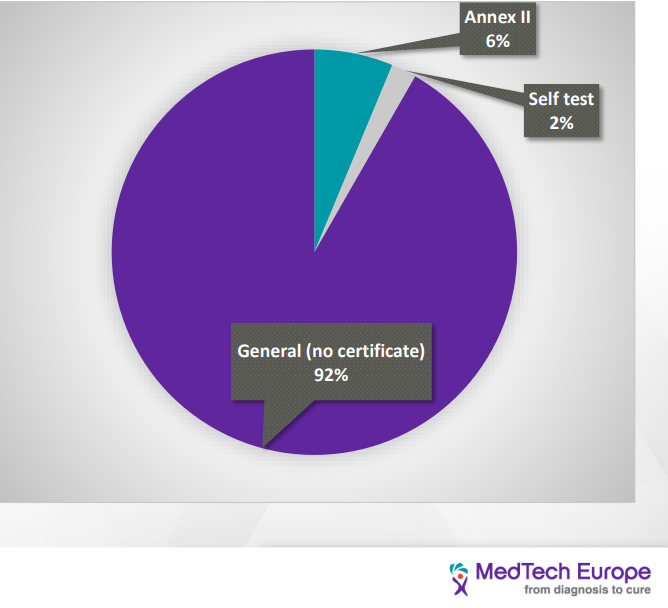
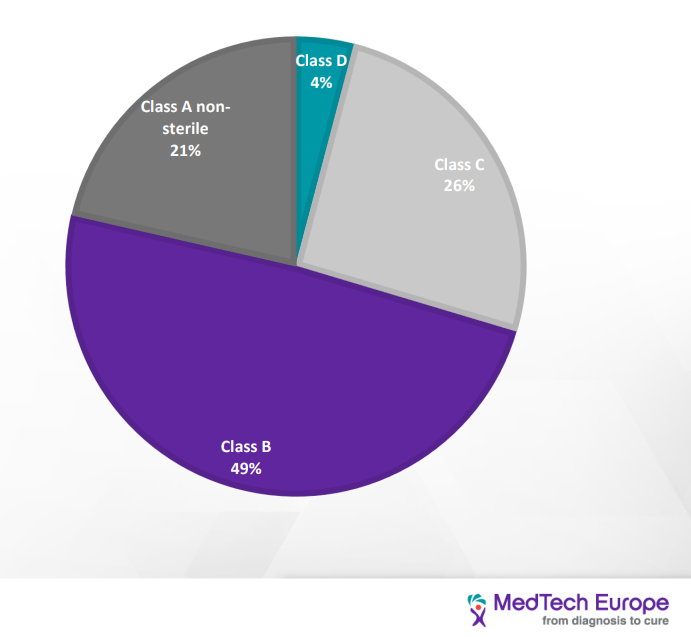
IVDD classification is “list-based” and divided into four categories: (from the lowest to the highest risk): General, self-test, list B (annex II), list A (annex II). IVDR is Risk-based and divided into four categories (from the lowest to the highest risk) Class A, A sterile, B, C, D
IVDR classification is based on seven rules; one should go over each rule and see if it applies to its device; the NB wants to know the process of classification, and so you need to address each rule that applies, then you indicate which is the highest class/ highest risk and that’s the rules that apply.
As demonstrated in the picture above,there is a distinct correlation between the class of the device (risk) and the degree of regulatory requirements and the NB monitoring.
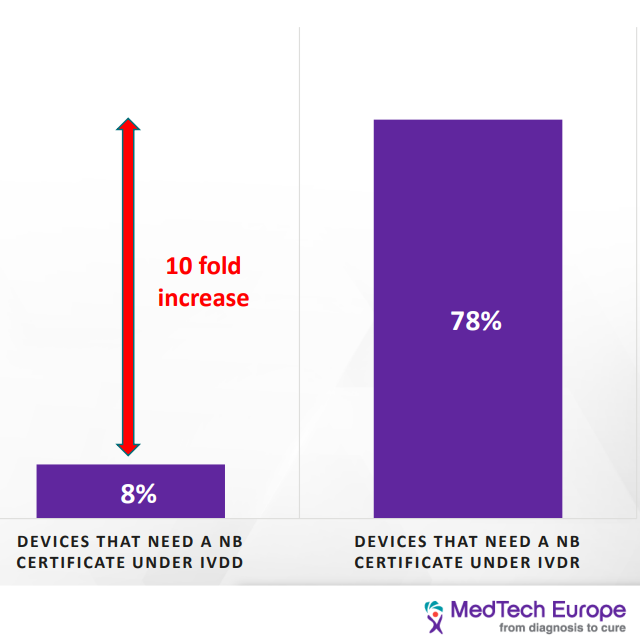
This change in classification has a significant impact on most IVD devices. Under the IVDD, there are about 40,000 devices. Of them, only 8% require NB certification; however, it is estimated that under the IVDR classification, 78% of the devices would need NB certification. Furthermore, it is estimated that about 22% of devices will not seek accreditation under IVDR due to the increased cost. [2] Products sold under FDA approval may find that they have a leg up in gathering the necessary information.
Distribution of IVD devices based on the IVDD classification
Distribution of IVD devices based on the IVDR classification
As we can see, the load on the NB is going to increase tenfold due to the increasing number of devices that would seek certification, and at the same time, the number of NBs qualified to provide certificates for the IVDR has decreased from twenty-one to six. This would have a significant impact on the NB availability and timeline to process new devices. Current turnaround time is 9-11 months and go as long as 18 months. [3]
The change in classification method will drastically change the class of many devices, as demonstrated in the illustration above and by so, also the number of regulatory requirements to comply with. For example, a COVID-19 self-test is classified as list B under the IVDD. In contrast, under the IVDR, it will be class D. Another example is the COVID-19 PCR test that under the IVDD is self-certified were as under the IVDR, it is class D (highest risk category).
The best practice is first to figure out the new classification of one’s device, which will allow you to know the regulatory requirements, perform a gap assessment, and create a plan to close the gaps.
Clinical Evidence/ Performance Evaluation
Since the IVDs don’t have direct contact with the patient and don’t impact the patient’s health directly, there is no clinical evaluation but a performance evaluation.
BSI Data revealed that 40% of the first round of questions were about clinical performance. The next topic was in regards to analytical performance. [4]
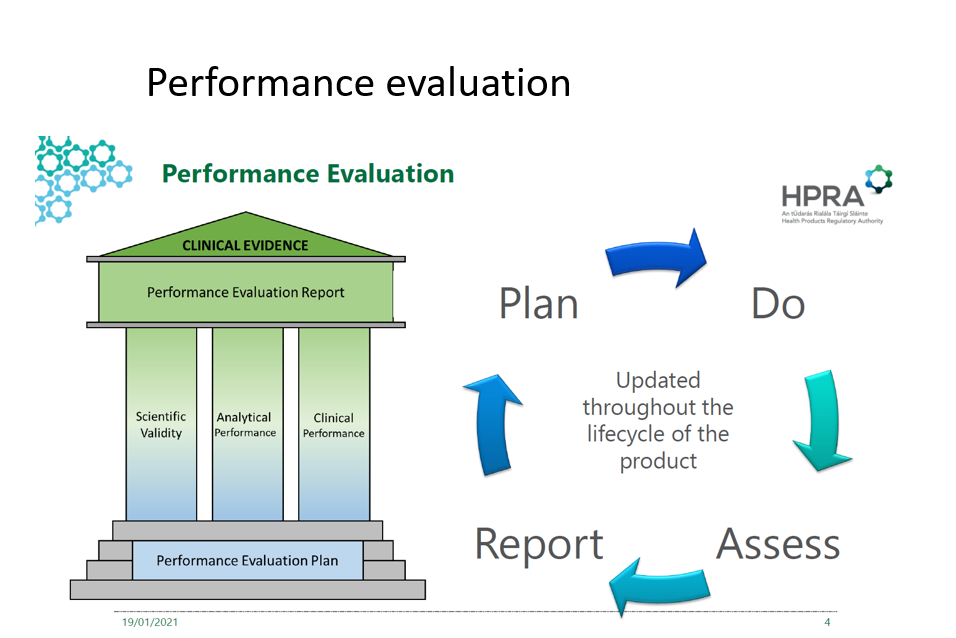
The performance evaluation for IVD is composed of three pillars:
Scientific validity- means the association of an analyte with a clinical condition or a physiological state; 2017/746 (38)
Analytical performance- means the ability of a device to correctly detect or measure a particular analyte; 2017/746 (40). This may be demonstrated with the following test: sensitivity, specificity, precision, accuracy, the limit of detection, cut off, etc.
Clinical performance- The ability of a device to yield results that are correlated with a particular clinical condition or a physiological or pathological process or state in accordance with the target population and intended user. 2017/746 (41)
The picture above demonstrates the continuous cycle of performance evaluation and its pillars.
State of the Art: Means what is currently regarded as good practice, not the most advanced technology [ISO/IEG Guide 63:2019, 3.18]
Performance evaluation aims to scientifically demonstrate that the device is safe, state of the art and that the intended clinical benefit is achieved. This can and should be done by relevant study data, literature, and other sources of technical information and the analysis and conclusions of the performance evaluation process.
Seen as the new regulation has put a big emphasis on the whole lifecycle of the device, the manufacturer is required to demonstrate the info for the certification and then continuously and actively gather this information throughout the whole lifecycle of the device, together with the post-market surveillance (PMS).
A few tips that we learned along the way:
● Start sooner rather than later; it’s a long, unfamiliar process, and the queue is getting longer by the minute.
● The NB wants to see the IVDR jargon used.
● Under the IVDR, you only get three rounds of questions from the NB, and then you have to restart (Unlike in the IVDD, where you could go back and forth with the NB)
● Whenever you don’t meet a requirement since it’s not applicable, you must provide a justification
● Provide an extensive glossary and acronyms (the reviewer may be a health professional, but not necessarily from the industry).
● Make sure your crucial narrative/stories are clear, that each document has a beginning, middle, and end, and summaries of the conclusions.
●Be consistent! Review for consistency in the document and across all the documents for the same names, terms, definitions, etc.
This article was prepared by:
Dr. Tami Siniaver
RA & QA Project Manager, Medical device
For more information about our medical device industry, visit:
During the past two years, we all were forced to adapt our quality activities, especially, when traveling became difficult and face-to-face meetings became limited. Particularly challenging during the COVID-19 Pandemic are supplier quality audits, an essential part of a supplier qualification program, in accordance with current regulatory requirements.
Advanced informational technologies available today, allow for effective and successful remote audits. For instance, live stream or pre-recorded video can be used for the examination of facilities and operators. Video conference services are widely used for group meetings and can be used for other purposes, such as operator interviews, screen sharing of documents, data, and other relevant information. In addition, confidential documents and data can be shared via different secure share folder services.
With the growing hope that the world will be back to normal very soon, we also want to believe that the lessons and experience gained during the last two years will be useful in the future. For example, remote supplier audits can be used also after the Pandemic is over. However, the advantages and disadvantages of remote supplier audits should be well recognized before deciding on which type of audit to plan.
Remote audits advantages:
Remote audits have numerous advantages over on-site audits for both auditors and auditees.
● Firstly, by default, they are much easier to schedule and allow more flexibility for both sides. For example, relevant auditee experts can join the discussions for a limited time, even if their general availability is low.
● Secondly, remote audits save resources and time, allowing for a more efficient audit program. Reduced travel and accommodation fees for the auditors, enables scheduling more than one vendor audit per week.
● Thirdly, remote audits may even be more efficient than on-site audits, if documents are shared by the auditees in advance since more documentation can be reviewed. If agreed in advance between both sides, video and audio recordings of the audit can provide better context for the paper records and allow improved follow-up for auditor and auditees
Remote audits challenges:
Remote audits challenges,that should be taken into consideration, while planning a remote audit.
● Firstly, despite all the technological solutions, remote audits provide limited ability to observe the facilities, equipment and activities. The level of detail that can be noticed remotely is relatively low and it is possible that the auditor will miss important details. Also, remote conversations are prone to misconceptions, which can be avoided by direct communication.
● Secondly, the remote facility tours require adequate infrastructures, such as sufficient internet coverage in all areas, which may be challenging, for example, in underground facilities or other remote areas. In addition, the personnel who are participating in the audit from both sides should be familiar with all remote access and data security technologies, such as document exchange, virtual chats, access permissions, and others. Lack of adequate infrastructure and personnel training will most probably lead to an unsuccessful audit.
● Thirdly, despite more flexibility with remote audits, different time zones can be a critical issue. Inconvenient audit hours may lead to reduced attention, lack of cooperation, frustration, and in general, a less successful audit.
● Lastly, we should always remember and as far as possible, prepare for the risk for technology failures, that may occur during the remote audit from either side, including sudden loss of connectivity, poor audio and/or video connection, and others.
In conclusion, on-site inspections will likely continue to be the preferred standard in the foreseeable future. Even the most successful remote audit may not be sufficient for supplier qualification and an on-site visit will yet be required to complete the qualification. Nevertheless, remote audits can be used in the supplier qualification process, as a valuable part of the general qualification program.
Remote supplier audits recommendations for successful outcome:
In order to maximize the effectiveness of the remote audit we have the following recommendations.
●The focus of the remote audit should be on the overall product quality/safety maintenance, general quality system, and critical operations, rather than on particular details.
● Sufficient technological infrastructure should be in place to support secure remote access, including internet coverage, suitable software for data exchange, and video conference. The participating personnel should practice virtual presentation skills and receive sufficient training to resolve common technical problems that may occur during the audit.
● An on-site follow-up should be considered to complete the detailed process observations and clear up any remaining issues.
We wish our colleagues fruitful and interesting audits, on-site or remote, and hope our recommendations will be useful.
This article was prepared by:
Orit Gamburg, B.Pharm, M.Sc., MBA
Gsap RA & QA Project Manager
For more information about our advanced therapies industry visit:
The continually-evolving field of medical cannabis in Israel poses challenges on medical cannabis companies and start-ups both due to regulatory ambiguity and due to differences in requirements between countries. By constantly updating regulatory changes and through its range of services, Gsap supports and accelerates dozens of medical cannabis projects in Israel and around the world, including the construction of post-harvest and cannabis production facilities, development of cannabis-based treatments and medical devices, and clinical trials of cannabis-based products.
As part of the actions to promote research and development of cannabis in Israel, applications for licensing and conducting research in the field of cannabis can be submitted to the IMCA as detailed inProcedure 108– Guidelines for submitting applications for licensing research in the field of cannabis.
The use of cannabis for medical purposes has accelerated in recent years in Israel and many countries around the world. In parallel, there is significant progress in scientific research in the field of cannabis for the purpose of establishing it as a plant with a beneficial effect on a variety of medical conditions. However, due to the status of the cannabis plant as an illegal substance in many countries, there is still a significant lack of evidence-based medical knowledge (EBM) about the mechanism of action of its active ingredients in the human body and about medical indications that can benefit from it.
Promoting evidence-based medicine and encouraging scientific research in the cannabis field are guiding principles in the reform of cannabis medicalization in Israel. Israel is one of the leading countries in the field of cannabis’ research and development and currently holds many studies in various areas: from cannabis plant science, agrotechnology, medical cannabis product development to clinical trials in humans.
The Cannabis Research and Development Committee has been appointed to examine research applications to meet regulatory requirements and consists of a wide range of experts from the fields of regulation, agriculture, life sciences, chemistry, and medicine. The committee meets 4 times a year, once a quarter. Requests for research are discussed by the committee in the order in which they are received. The IMCA makes every effort to discuss all the applications received by the committee close to the date of submission, but it is possible, depending on the submission date, that the application will be forwarded to one of the following meetings.
Research licensing in the field of medical cannabis
Submitting applications for licensing research in the field of cannabis are detailed in IMCA Procedure 108 (last updated on May 2019).
Cannabis research for medical use licensing process involves three stages:
Stage 1: Submitting an application for a feasibility permit for the cannabis research protocol
The application should include the study protocol describing the study background and rationale from the literature, study protocol and methods, study analysis, timelines, measures of Cannabis protection, amount and frequency of use, and additional details depending on the specific research.
Stage 2: Submitting a request for revocation of information that prevents issuing of a license and security clearance
Following approval of the study protocol by the committee, the sponsor must submit information about all stakeholders and employees involved in the cannabis research.
In addition, the research site is required for approval of its security conditions in accordance with IMC-GSP regulation.
Stage 3: Submitting an application for a licensing permit and research that includes contact with cannabis
Following obtaining a security approval for each of the research site, a final formal application will be submitted to the IMCA unit to grant the final authorization for research work with medical cannabis.
Recent updates in the medical cannabis field
● Quality requirements for the production of medical cannabis products – update April 2021 (Procedure 152)
Procedure 152 details the requirements and rules pertaining to the proper production and quality of medical cannabis products. This regulation details the following sections: Medical cannabis manufacturing regulation
● IMC-GMP authorization requirements and regulation
● Medical cannabis testing and labeling requirements
● Medical cannabis specifications
According to procedure 152, cannabis products authorized for production and consumption in Israel must be of the following forms: Medical cannabis inflorescence for smoking
● Medical cannabis oil extract for sublingual administration
● Medical cannabis cookies- for oral consumption
Per each of the forms, there is a range of concentrations of active ingredients (CBD, THC, and CBN) in cannabis products allowed for production and distribution.
Cannabis products, either in other configurations or in a different concentration of active ingredients may be distributed and consumed only after the applicant has demonstrated the safety and effectiveness of the product and it was approved by the IMCA.
On April 2021 an update was issued by the IMCA regarding additional categories of cannabis products approved for production according to IMC-GMP quality procedures [1]
Only cannabis products listed in the table including their API concentration ranges are approved for manufacture, distribution, and medical use. It should be emphasized that all approved cannabis products should be grown, produced, undergo laboratory tests for batch release, transported, stored, and issued only in accordance with the quality requirements of the IMCA as specified in procedures 152 and 154.
Approval of new indications for treatment with medical cannabis
Procedure 105 (updated on May 2019) details the procedures for the Cannabis indication committee. This committee deals with expanding/reducing the number of indications, clinical recommendations, examining ethical rules and pharmacovigilance rules.
In order to approve a new indication for cannabis, a sponsor or a principal investigator must submit a clinical trial application to the Ministry of Health (MOH). The clinical trial must include a sufficient number of patients to draw a conclusion regarding the safety and efficacy of cannabis for treatment in the required indication. A PK results may be warranted based on the specific indication. Due to the wealth of published information related to cannabis safety, preclinical studies are usually not a prerequisite for clinical trials.
Based on a successful clinical trial with a new indication or new formulation, the IMCA 154 and 152 procedures might be updated to capture the new indication or new formulation.