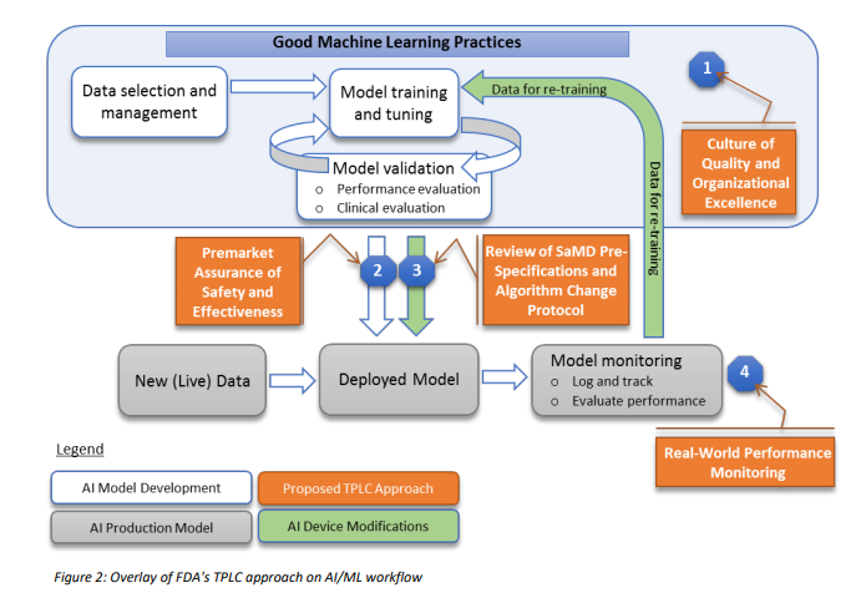
The United States Food and Drug Administration (FDA), the European Commission, and other regulators have made patient safety a strategic priority, by developing UDI regulations for Medical Devices and In Vitro Diagnostic (IVD) Devices, and are aiming for a globally harmonized and consistent approach, aligned with the IMDRF (International Medical Device Regulator Forum) Guidelines.
Topics in this article:
- What is the UDI?
- What is the purpose of UDI?
- UDI in the US
- FDA UDI Compliance Dates
- EU MDR UDI
- Implementation of the UDI system
What is the UDI?
The UDI (Unique Device Identification) is a series of numeric or alphanumeric characters that is created through a globally accepted device identification and coding standard. It allows the unambiguous identification of a specific medical device on the market.
The UDI may consist of plain text (human-readable) and AIDC (machine-readable) and is composed of two parts:
- Device Identifier (DI) – A unique numeric or alphanumeric code specific to a device version or model. The Device Identifier (DI) is the first part of a UDI.
- Production Identifier(s) (PI) – Numeric or alphanumeric codes that identify production information for a device and can include the following:
- Lot or batch number;
- Serial number;
- Expiration date;
- Manufacturing date;
- Distinct identification code (for Human Cell and Tissue).

- Reprinted from https://www.reedtech.com/
What is the purpose of UDI?
The UDI system facilitates easier traceability of medical devices, significantly enhances the effectiveness of the post-market safety-related activities for devices and allows for better monitoring by competent authorities. It also helps to reduce medical errors and to fight against falsified devices. The use of the UDI system finally should also improve purchasing and waste disposal policies and stock management by health institutions and other economic operators.
Which products are subject to the UDI system?
The UDI system should apply to all medical devices, except custom-made and performance study/investigational devices.
UDI in the US
The US Food and Drug Administration (FDA) released in September 2013 a UDI rule which establishes a UDI system applying to all medical devices placed on the US market.
The Unique Device Identification System final rule (UDI Rule) requires device labelers (typically, the manufacturer) to:
US UDI requirements
- Include a unique device identifier (UDI) on device labels and packages, except where the rule provides for an exception or alternative (a UDI is not required to be placed on any shipping container (logistics unit)).
- If a device is intended for more than one use and intended to be reprocessed before each use, the device labeler must also mark the UDI directly on the device.
- Submit device information to the Global Unique Device Identification Database (GUDID), i.e. the US FDA UDI regulatory database.After receiving a DI and a PI nomenclature from an agency, companies must register the DI in GUDID. The database does not store PI information, as this generally changes from batch to batch.
How to obtain a US UDI?
There are currently three agencies approved by FDA to issue UDIs: GS1, HIBCC, and ICCBBA. Each agency has its own labeling system. All three agencies issue Device Identifiers and nomenclatures for creating Production Identifiers, the main components of a UDI.
While the UDI is created through the guidelines of an approved issuing agency, the medical device manufacturer is responsible for performing a submission of the identifier along with several product data attributes.
FDA UDI Compliance Dates
FDA established a staggered series of compliance dates based on the medical device risk classification system.

- Reprinted from https://medicaldeviceacademy.com/
* Final guidance from the US Food and Drug Administration pushes back enforcement deadlines for certain Unique Device Identification (UDI) requirements for Class I and unclassified medical devices due partially to the agency’s coronavirus pandemic-related priorities.
According to the final guidance, FDA will delay enforcement of UDI labeling, date formatting as well as Global Unique Device Identification Database (GUDID) submission requirements for Class I and unclassified devices until September 24, 2022. Enforcement of compliance deadlines for these requirements had previously been set for September 24, 2020.
EU MDR UDI
The European Commission has also developed UDI requirements that are part of the EU Medical Devices Regulation (MDR) and the In-Vitro Diagnostics Regulation (IVDR). The European Union Medical Device Regulation (EU MDR) Articles 27, 28, and 29 and Annex VI of the regulation cover UDI.
EU MDR UDI requirements
- Full UDI on Device Label and packaging, presented in human-readable plain text and Automatic ID and Data Capture (AIDC) technology, e.g. 1D/2D barcode, RFID (shipping containers shall be exempted from the UDI requirement)
- Permanent UDI marking on reusable devices
- Submit device ID and attributes to the EUDAMED database, i.e. the EU regulatory database for regulated medical devices.
- Reporting requirements: include UDI in annual reports, post-marketing surveillance and the Patient Implant Card
The EU MDR uses the concept of “Basic UDI-DI.” This is general code for a product line or set of related models that have the same intended use. The Basic UDI-DI is separate from the UDI-DI, which is a unique DI that is specific to a particular model.
The Basic UDI-DI is not required on labeling or the device itself but is used in Certificates of Conformity, Summaries of Safety and Clinical Performance, Eudamed submissions, and other documentation.
EU MDR UDI database
The Eudamed database will store EU UDI information. Manufacturers/Authorized Representatives/Importers will be required to keep records of all UDIs assigned for their devices. Companies are strongly encouraged to store their UDI information electronically and back it up in multiple locations because the EU UDI requirements go into effect before the Eudamed database comes online in 2022. Parts B and C of Annex VI of the regulation cover the information required.
How to obtain an EU UDI?
There are currently four agencies approved by the EU to issue UDIs: GS1, IFA GmbH, HIBCC, and ICCBBA.
Deadline for a medical device to comply with the EU UDI requirements
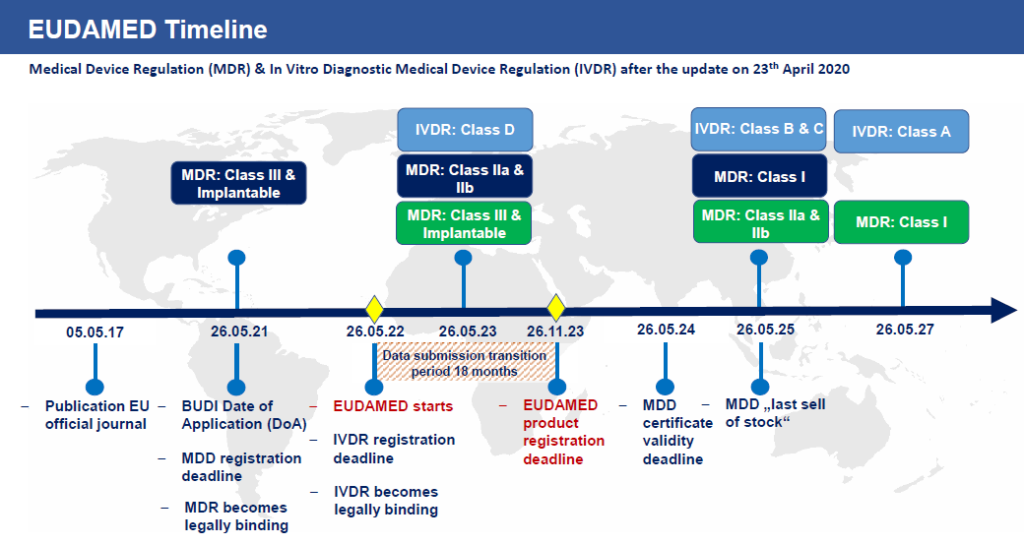
- Reprinted from https://www.europe-it-consulting.ch/?lang=en
Implementation of the UDI system
The general steps for establishing the required UDIs are outlined below:
- Determine which products/configurations/accessories/kits require UDI
- Select and register with the accrediting agency of your choice
- Obtain the necessary DIs from the accrediting agency
- For each DI, determine what PI information will be included based upon the product labeling
- Select your barcode format and modify labeling as required to accommodate
- Prepare the necessary infrastructure to support the UDI process, including:
− Updates to quality system processes to ensure DIs are created and maintained properly
− Updates to label creation, printing, and inspection processes
− Installation and validation of label printing equipment as needed
In parallel, you must register with the relevant UDI databases.
Gsap experts have the expertise and resources to help you establish compliance processes to meet UDI requirements, and will be happy to assist you to create the right UDI label solution for your medical device.
Liat Aharon Pollak,
RA & QA Project Manager